










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkBiocell
Print version ISSN 0327-9545
Biocell vol.31 no.2 Mendoza May/Aug. 2007
p53-Rb signaling pathway is involved in tubular cell senescence in renal ischemia/reperfusion injury
Li Kailong1,2, Du Xiaolan1, He Yani2, Zhao Lin1, Yang Jvrong2, Song Ruihua1, And Chen Lin1.
1 Center of Medical Molecular Genetics, State Key Laboratory of Trauma, Burn and Combined Injury;
2 Department of Nephrology, Daping Hospital, Third Military Medical University, Chongqing 400042, China.
Address correspondence to: Dr. Li kailong. Department of Nephrology, Daping Hospital, 10 Changjiang Zhi Road Daping, Chongqing 400042, CHINA. Fax: +86-23-68710162. E-mail: likailong1966@163.com
ABSTRACT: Objective: To investigate the course of tubular cell senescence and expressions of p53, p21, and Rb during the late phase of ischemia/reperfusion (IRI) in the kidney, and assess the effects of the p53-Rb pathway on tubular cell senescence. Methods: Experimental models of unilateral renal IRI were used in p53(+/+) and p53(-/-) mice. Histological changes at the tubular level, progress of cell senescence, and the expression of Rb, p21, and/or p53 proteins in tubular cells were studied at different moments in time after IRI. Results: Chronic tubulointerstitial fibrosis was much more severe and widely distributed in IRI kidneys of p53(+/+) mice in later stages than in earlier stages. Senescent tubular cells were significantly increased at 3 and 6 months after IRI. In contrast, in contralateral kidneys of p53(+/+) mice and in both kidneys of p53(-/-) mice, almost no senescent cells were observed at 1 and 3 months after IRI, and only a few senescent cells were detected in IRI kidneys of p53(-/-) mice at 6 months. In mice of both genotypes, cell senescence was correlated with the expression levels of p53, p21, and Rb proteins. Conclusion: The IRI accelerated tubular cell senescence is presumed to be one of the mechanisms of the "long-term effect" of IRI. Furthermore, the activation of p53-Rb signaling pathway may play a vital role in tubular cell senescence induced by IRI.
Key words: Cell senescence; Cell cycle regulatory protein; Ischemia/reperfusion injury; Kidney.
Introduction
Ischemia/reperfusion injury (IRI) plays a major role in delaying graft function and the long-term changes after kidney transplantation (Gueler et al., 2004; Es et al., 1983). One of the theories to explain the progressive deterioration of IRI kidney function was that of replicative senescence of the renal tubular epithelial cell. Senescence of an organ or tissue results from aging and/ or environmental stress-dependent modification of cellular function, and, with time, the accumulation of cellular alterations may lead to deleterious effects in various organs and tissues and adversely affect IRI kidney function Chkhotua et al., 2002). It has been well documented that the kidney develops characteristic physiologic and pathologic changes with aging, which includes cortical volume loss, fibrous intimal thickening of the arteries, glomerulosclerosis, tubular atrophy, and substantial function decline (Epstein, 1996; Goyal, 1982; Linderman et al., 1985). In general, these changes are called 'senescence' and are associated with significant clinical problems. Elderly people, for example, are up to 100 times more likely to develop end-stage renal disease (ESRD) than young people. Donor's age has been proven to be one of the main predictive factors for graft function recovery and survival (Chkhotua et al., 2002). Chronic allograft nephropathy (generally caused by IRI) has been found to share common pathologic changes with an aging kidney (Halloran et al., 1999). A recent study showed that renal ischemia, followed by concordant haemoperfusion, is associated with kidney telomere shortening and the over-expressions of p21 and p27 CDKI genes in the kidney. Ischemia during transplantation results in telomere shortening and the subsequent activation of p21 and p16, and senescence-associated b-galactosidase (SA-b-gal) staining has also been observed in chronically rejected kidney grafts, which indicates that shortening of telomeres is not the sole reason for cells to become SA-b-gal-positive (Chkhtua et al., 2005; Joosten et al., 2003). The senescent renal tubular epithelial cell is a strong marker of an aged kidney and is associated with chronic renal pathological changes (Chkhotua et al., 2002; Joosten et al., 2003; Chkhotua et al., 2001; Meguid et al., 2005; Ding et al., 2001; Melk et al., 2004). Therefore, the mechanism of cell senescence caused by IRI is relevant to chronic kidney disease and chronic allograft nephropathy. Recent findings suggest that p53-Rb and p16-Rb pathways cocontribute to overall cell senescence, which is caused by extrinsic and intrinsic stimuli (Hasty and Vijg, 2004; Ben-Porath and Weinberg, 2005; Campisi, 2005a, b; Famulski and Halloran, 2005; Taylor et al., 2004), and the expression of cell cycle regulatory proteins, such as p53, p21, and Rb, are showed to be up-regulated during the initiation and extension phase of IRI (Chkhotua et al., 2005; Famulski and Halloran, 2005; Kelly et al., 2003; Devarajan et al., 2003; Megyesi et al., 2001; Price et al., 2004; McLaren et al., 2004). However, the expression of p53, p21, and Rb and the senescent progression of renal tubular epithelial cells in late phase of IRI, as well as the underlying mechanism, have not yet been assessed.
The aim of the this study was to determine the course of tubular cell senescence and the expression of some cell cycle regulatory proteins in the kidney in the late phase of IRI and to assess the effects of the p53-Rb pathway on tubular cell senescence. For this purpose, SA-b-gal activity and the expression of p53, p21, and Rb at 1, 3, and 6 months after IRI were studied with an experimental model of unilateral renal IRI, in both wildtype [p53(+/+)]]]and p53 deficient [p53(-/-)] mice. This study provides the first report on the molecular basis of tubular cell senescence in the late phase after kidney IRI with the animal model of p53 deficient mice.
Materials and methods
Animal procurement and treatment
p53(-/-) mice (F2) in C57BL/6J mice (F2) were obtained from NIH, and housed in a SPF environment. The p53(+/-) mice were intercrossed with p53(+/-) mice to generate homozygous p53(-/-) mice and p53(+/+) littermates. Genotyping of the mice was performed by PCR analysis of DNA extracted from tail tips as reported by LA Donehower (Donehower et al., 1992). Two-monthold male p53(-/-) and p53(+/+) mice were used in this study. All animals were treated in a humane manner.
Renal ischemia/reperfusion injury model
We employed a well-established murine model of renal IRI, for which the structural and functional consequences in periods of renal ischemia have been extensively documented (Supavekin et al., 2003; Nogae et al., 1998; Daemen et al., 1999; Kelly et al., 2001). Male mice weighing 19 to 24 g were housed with a 12hour light and 12-hour dark cycle and were allowed unrestricted access to food and water. All animals were anesthetized with sodium pentobarbital (50 mg/kg i.p.) and then placed on a warming table to maintain a rectal temperature of 37ºC. The left kidney was exposed by a flank incision, then the left renal pedicle was occluded with a nontraumatic vascular clamp for 45 min, during which the kidney was kept warm and moist. The clamp was removed to return the blood flow to the left kidney, and the incision was sutured. The mouse was allowed to recover in a warm cage. At 1, 3, and 6 months after reperfusion, the mouse was sacrificed with intraperitoneal pentobarbital, and both kidneys were harvested (the right kidney served as a control for each animal). Five separate animals were examined at each reflow period. Each kidney was slit by center sagittal section into three parts medially for the following analysis.
Morphologic studies of tubulointerstitial injury
Paraffin-embedded sections of 4 mm were prepared and stained with hematoxylin and eosin, and then were examined in a blinded manner by two examiners, with each section evaluated twice. Tubulointerstitial injury was defined as tubular atrophy, dilation, and intratubular casts, as well as thickening of tubular basement membranes, cellular infiltration, and widening of the interstitium, which was scored semi-quantitatively according to the method of Shih et al. (1988) as follows: 0 (normal), 0.5 (small focal area injured), 1 (less than 10% of the cortex injured), 2 (10% to 25% of the cortex injured), 3 (25% to 75% of the cortex injured), and 4 (more than 75% of the cortex area injured).
Senescence-associated -b-galactosidase (SA-b-gal) assay in situ
Cryostat sections (4 mm) of the kidney were mounted onto glass slides and fixed in 0.5% glutaraldehyde in PBS at room temperature for 10 min. The sections were washed in PBS and incubated at 37ºC (no CO2) with fresh 5-bromo-4-chloro-3-indolyl-b-Dgalactopyranoside (code: GALS, X-gal, Sigma, St.Louis, MO) staining solution, according to Dimri et al. (1995): 1 mg of X-gal (stock, 20 mg/ml of dimethylformamide, Sigma)/ml of 5 mM K3 Fe(CN) 6 , 5 mMK4Fe(CN)6, 150 mM NaCl, 2 mM MgCl2 in 40 mM citric acid/sodium phosphate, pH 6.0. Tissue sections were examined after incubation for up to 14 hours and counterstained with eosin. A set of slides was produced and photographed, and then quantification of the SA-b-gal staining was accomplished by Image-Pro Plus Software (5.0.2). The image was opened in Image-Pro Plus, and staining density for the whole section was calculated. The mean staining density of five independent experiments was taken for further calculation and statistical analysis (Melk et al., 2003).
Immunohistochemistry for p53, p21, and Rb
Immunostaining for p53, p21, and Rb was performed on cryostat sections (4 mm) by using the standard avidin-biotin complex method. Sections were fixed with 4% formaldehyde/PBS (pH 7.4) and treated with 3% H2O2 in methanol for 10 min to inactivate endogenous peroxidase. After washed in PBS, the sections were microwaved in 10 mM citrate buffer (pH 6.0) for 10 min to retrieve the antigen. Sections were then incubated with 1.5% normal goat serum for 15 min, and then incubated with primary antibodies, either rabbit anti-mouse p53 polyclonal antibody (1:200, clone FL-393, code: SC-6243, Santa Cruz Biotechnology, Santa Cruz, USA), rabbit anti-mouse p21 polyclonal antibody (1:200, clone M-19, code: SC-471, Santa Cruz Biotechnology, Santa Cruz, USA), or rabbit anti-mouse polyclonal antibody against total retinoblastoma (1:200, clone C-15, code: SC-50, Santa Cruz Biotechnology, Santa Cruz, USA) at 37ºC for 3 hr. After removal of unbound primary antibody and rinsing with PBS, sections were incubated with avidin-biotinylated horseradish peroxidase (code: pk-7200, Vectastain Elite ABC kit; Vector Laboratories) for 60 min. The slides were washed again in PBS, visualized using the diaminobenzidine (DAB) substratechromogen system (code: 3468, Dako, Glostrup, Denmark), and counterstained with haematoxylin. Finally, sections were washed with tap water, dehydrated, and mounted. Negative controls consisted of substituting the primary antibody with PBS. To evaluate the expression of each cell cycle regulatory protein, two examiners per animal counted the number of nuclei that were positively stained. Examiners used 20 randomly chosen highpower eye grid fields (HPF).
Western blot analysis for p53, p21, and Rb
Pieces of dissected renal tissue were immersed in lysis buffer (50 mM Tris-HCl, pH 7.4, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 100 mM NaCl, 0.1 mM sodium orthovanadate, 1 mM sodium fluoride, 10 mg/ml aprotinin, 10 mg /ml leupeptin, 10 mg /ml pepstatin, and 10 mg /ml PMSF), homogenized with a dounce homogenizer, and then incubated on ice for 30 min. After centrifuging the mixture at 20,000 g 4ºC for 30 min, the protein concentration of the collected supernatant was determined by using a BCA kit (code: 23250, Pierce, Rockford, IL). Samples of 40 mg protein were mixed with 2 ¥ loading buffer containing 125 mMTris-HCl (pH 6,8), 5% glycerol, 2% sodium dodecyl sulfate (SDS), 2% bmercaptoethanol, and 0.001% bromophenol blue and were electrophoresed on 7.5% sodium dodecyl sulfate polyacrylamide gels for Rb or on 10% gels for p53 and p21. A biotinylated protein ladder (code: 7727, Cell Signaling Technology) was used in each gel. The proteins were transferred overnight to polyvinylidine difluoride (PVDF) membranes (Millipore, Bedford, MA) using a Bio-Rad Western blotting apparatus. After the proteins were transferred, blots and gels were stained with Coomassie blue to check for complete protein transference and equal loading. Membranes were blocked in 5% skim milk and hybridized to the following primary antibodies against p53, p21, and Rb, as mentioned above (1:2000), and mouse actin monoclonal antibody (code: Ab-5, LabVision Corporation, USA) 1:2000. Membranes were washed in 20 mM Tris, pH 7.5, 150 mM NaCl, 0.05% (v/v)Tween-20, and hybridized to the corresponding horseradish peroxidase-conjugated secondary antibodies, goat anti-rabbit IgG-HRP (code: SC-2004, Serologicals Corporation) 1:3000, or goat anti-mouse IgG-HRP (code: 12-349, Serologicals Corporation) 1:3000 at room temperature for 1 hr. Chemiluminescence detection was performed using Supersignal (code: 34080, Pierce, IL, USA) according to the manufacturer's instructions, and images were acquired on X-ray film. Quantitative densitometry was performed on the identified bands using a computer-based measurement system.
Statistical analysis
Data analysis was performed using SPSS (SPSS Inc, Chicago, IL, Version 11.0). The means of the tubulointerstitial fibrosis index score (histological index score) among the three moments in time (1, 3, and 6 months after kidney IRI) in both groups [p53(+/+) mice and p53(-/-) mice] were compared using KruskalWallis non-parametric tests. The Fisher exact test was used to determine the differences in the expression level of the cell cycle regulatory proteins in both groups and at different moments in time after IRI. Linear correlation analysis was used to calculate the correlation between the SA-b-GAL activity and the expression of each cell cycle regulatory protein. Statistical significance was defined as P<0.05 and is indicated in the text.
Results
Histopathology of tubulointerstitium
In all of the p53(+/+) and p53(-/-) mice, the morphology of the tubulointerstitium was normal in the right kidneys at 1, 3, and 6 months after IRI (pictures are not shown). But the appearance of abnormalities, including intratubular casts, tubular dilation and atrophy, intertubular cell infiltration, and matrix accumulation, occurred in the left kidneys at 1 month, and these abnormalities were progressively aggravated at 3 and 6 months. Furthermore, the tubulointerstitial injury was much more severe and diffused in p53(+/+) mice than in p53(-/-) mice (pictures are not shown). The semiquantitative analysis of tubulointerstitial fibrosis in p53(+/+) and p53(-/-) mice after IRI is summarized in Table 1.
TABLE 1.
Semiquantitative analysis of tubulointerstitial fibrosis in IRI kidneys of p53(+/+) and p53(-/-) mice*

SA-b-gal activity in the IRI kidney
SA-b-gal has been recommended as an indicator of cell senescence both in vitro (Bodnar et al., 1998) and in vivo (Ding et al., 2001; Sigal et al., 1999). Cryostat sections of IRI kidneys were histochemically stained for SA-b-gal activity, with indigo blue-colored pigment appearing at pH 6.0, to explore if senescent cells can be demonstrated in situ in the IRI kidney. In the IRI kidneys of p53(+/+) mice, positive staining for SA-b-gal activity in tubular segments began to appear occasionally at 1 month after IRI (Fig. 1A), and became increased obviously at 3 and 6 months after IRI (Fig. 1B and 1C). But, in contralateral kidneys of mice of either genotype at all specified times after IRI [Fig. 1D, p53(+/+) mice; 1H, p53(-/-) mice, both at 6 months] and in the IRI kidneys of p53(-/-) mice at 1 and 3 months (Fig. 1E and 1F), there was almost no positive staining; positive staining was only occasionally detected in partial segments of tubules in the IRI kidneys of p53(-/-) mice at 6 months (Fig. 1G). SA-b-gal staining of various degrees was found in mainly the corticomedullary portion of the IRI kidneys and was not detected in the glomeruli of all the kidneys at each time checked after IRI. SA-b-gal staining was quantified by using a photoimaging technique. Mean staining densities are shown in Table 2. As a control for the staining specificity, tissues were stained at pH 7.4, and b-galactosidase activity was not observed in any of the groups.

FIGURE 1. Senescence-associated b-galactosidase (SA-b-gal) staining. Frozen sections (4 um) of mouse renal cortex were fixed, stained, counterstained, and photographed randomly at 200xoriginal magnification. (A) In left kidney from p53(+/+) mouse at 1 month after IRI, there is occasionally positive staining for SA-b-gal in focal segments of tubules. (B and C) Left kidney cortices from p53(+/+) mice at 3 and 6 months after IRI show positive staining for SA-b-gal in partial and wholesegments of tubules with a increased frequency. (D and H) By contrast, in the contralateral kidneys from p53(+/+) mice and p53(-/-) mice at 6 months after IRI, there is almost no positive staining for SA-b-gal in tubules. (E and F) Left kidney cortices from p53(-/-) mice at 1 and 3 months after IRI have no positive staining. (G)In left kidney cortices from p53(-/-) mouse at 6 months, occasionally positive staining for SA-b-gal in focal segments of tubules was detected.
TABLE 2.
Quantitative analysis of SA-b-gal staining in IRI kidneys (Arbitrary units)
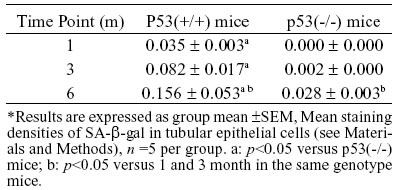
p53, p21, and Rb expression in IRI kidneys
Our semi-quantitative immunohistochemistry analysis (Table 3) demonstrated a higher levels of p53, p21, and Rb protein expressions in the IRI kidneys of p53(+/+) mice at 1, 3, and 6 months after IRI compared to the right kidneys (no IRI). However, in p53(-/-) mice, p21 and Rb expressions were delayed and were at lower levels than those in p53(+/+) mice (p<0.05), although their expressions were still at a higher level compared to the contralateral kidney (no IRI kidney), especially at 6 months after IRI. In both p53(+/+) mice and p53(-/-) mice, the expression of each of these cell cycle regulatory proteins in the IRI kidneys was at significantly higher levels at 6 months compared to 1 and/or 3 months after IRI (p<0.05). Enhanced expressions of these cell cycle regulatory proteins were localized in the nuclei of tubular epithelial cells, and were found in mainly the corticomedullary portion of the IRI kidneys.
TABLE 3.
Positively stained cells per HPF for p53, p21 and Rb respectively in renal tubular epithelium of IRI kidneys*
Quantitative analysis of these cell cycle regulatory proteins was performed using western blotting, and this analysis showed alteration patterns similar to those found by the semi-quantitative immunohistochemistry analysis (Figs. 2, 3, and 4). As shown in Figure 4, Rb protein displayed two bands, the inactive Rb (phosphorylated form) and active Rb (hypophosphorylated form). The phosphorylated form migrated more slowly than the hypophosphorylated form on SDS-PAGE gels (Chen et al., 1989; Schonthal and Feramisco, 1993). The ratio of active Rb to inactive Rb can be used to determine the activity of total Rb, and in the kidney tissue, which also showed a alteration pattern similar to that found by the semi-quantitative immunohistochemistry analysis.
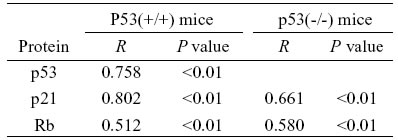
The statistical analysis indicated that changes of tubular cell senescence were correlated with the expression levels of p53, p21, and Rb proteins. The correlation coefficient (R) and the p value are shown in Table 4
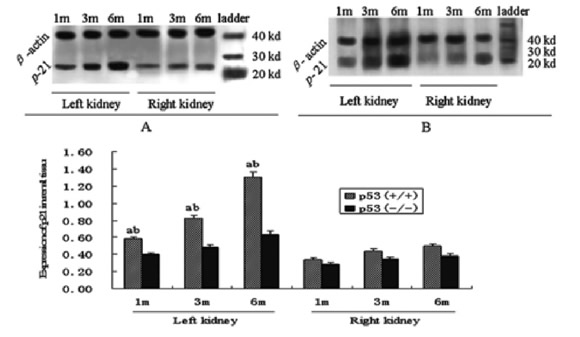
FIGURE 2. Expression of p21 in renal tissue of p53(+/+) (A) and p53(-/-) (B) mice after IRI. a: <0.05 versus p53(-/-) mice at the same time; b: p<0.05 versus right kidney at the same time and in the same genotype mice (Western blot analysis)
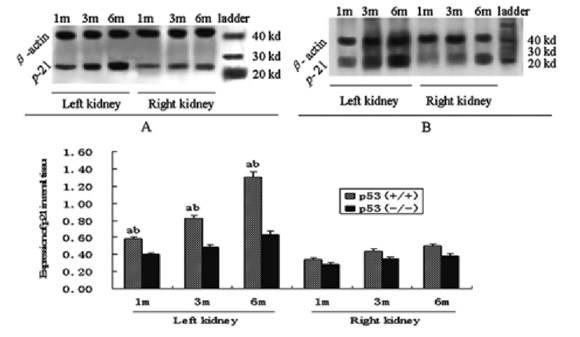
FIGURE 3. Expression of Rb in renal tissue of p53(+/+) (A) and p53(-/-) (B) mice after IRI. a: p<0.05 versus p53(-/-) mice at the same time, b: p<0.05 versus right kidney at the same time and in the same genotype mice (Western blot analysis)
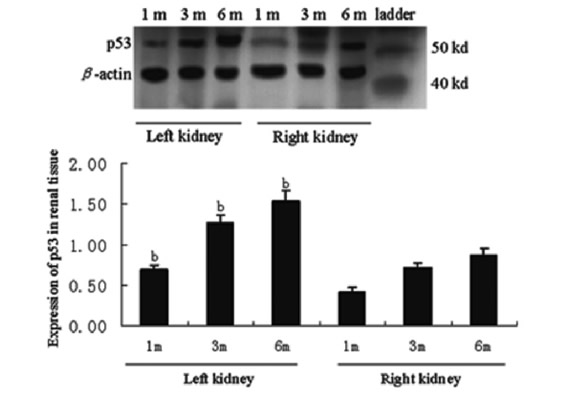
FIGURE 4. Expression of p53 in renal tissue of p53(+/+) mice after IRI. b: p<0.05 versus right kidneys at the same time (Western blot analysis)
TABLE 4.
Linear correlation between SA-b-gal activity and cell cycle regulatory protein in IRI kidneys
Discussion
Growing evidence indicates that renal vascular endothelial injury and dysfunction play an important role in initiating and extending renal tubular epithelial injury and, thus, contribute to the ongoing pathogenesis of acute renal failure caused by IRI (Molitoris and Sutton, 2004). In most cases, with the removal of harmful factors, the IRI is resolved, and kidney function is eventually restored (Haller et al., 1996). However, after initial recovery and a period of relative quiescence, in some cases, proteinuria and progressive morphologic changes, including glomerulosclerosis, arterial obliteration, and interstitial fibrosis may gradually develop. Although most studies have supported the concept that the inflammatory response is an important mediator of renal IRI and have shown that the inhibition of adhesion molecule expression, the decrease of endothelial cell activation, or the prevention of leukocytes adhesion can prevent or ameliorate delayed IRI kidney function, it is not clear why and how these acute effects may affect the long-term function of the IRI kidney (e.g, the transplanted kidney), and whether IRI plays a role in the development of chronic renal disease (e.g, the chronic allograft nephropathy). Obviously, the hallmark of chronic renal changes after IRI is fibrosis, and acute damages, such as necrosis and apoptosis, in the kidney after the initial injury may contribute to chronic fibrosis and deterioration. The loss of glomeruli and subsequent increase in "renal workload" have been proposed by others (Brenner and Milford, 1993; Azuma et al., 1997). Capillaries in other areas of the kidney may also be affected by IRI, and a rarefication of capillaries has recently been suggested as a critical step in the pathogenesis of chronic renal disease (Kang et al., 2002). However, the occurrence and mechanism of another type of cellular change, cell senescence, in pathogenesis of renal dysfunction in the late phase after IRI is not well understood. Cell senescence, especially tubular epithelial cell senescence, is associated with renal chronic pathological changes (Chkhotua et al., 2002; Joosten et al., 2003; Chkhotua et al., 2001; Meguid et al., 2005; Ding et al., 2001).The p53, p21, and Rb genes are correlated with cell senescence (Chkhotua et al., 2005; Joosten et al., 2003; Ben-Porath and Weinberg, 2005; Campisi, 2005a; Famulski and Halloran, 2005; Taylor et al., 2004), and the expressions of these cell cycle regulatory proteins can be up-regulated by some stressors, such as oxidative stress and DNA damage (Hasty and Vijg, 2004; Campisi, 2005b; Price et al., 2004; Melk et al., 2003; Cao et al., 2003; Tyner et al., 2002; Maier et al., 2004). In fact, oxidative stress and DNA damage be persistent in IRI kidneys, even in the late phase after IRI (Gueler et al., 2004; Melk et al., 2003; Khundmiri et al., 2004), which may result from the loss of glomeruli, the rarefication of capillaries, and chronic inflammatory cell infiltration in IRI kidney. We, therefore, propose that oxidative stress and/or DNA damage can induce the continuing activation of the p53/ p21-Rb pathway and eventually trigger and accelerate tubular epithelial cell senescence in the late phase of IRI. This may be one of the mechanisms responsible for the "long-term effect" of the IRI kidney. To test our hypothesis, we made an experimental model of unilateral renal IRI in p53(+/+) and p53(-/-) mice at 1, 3, and 6 months after IRI. We observed the histological changes of the renal tubule and assessed the senescence changes and the expression of Rb, p53, and/or p21 proteins in the tubular epithelial cells.
In this study, chronic tubulointerstitial changes were detected in IRI kidneys as chronic, progressive lesions, which included intratubular casts, tubular dilation, tubular atrophy, and thickening of the tubular basement membrane. These chronic changes were much more severe and diffused in p53(+/+) mice than in p53(-/-) mice and were present in later stages rather than in early stages after IRI. However, no obvious chronic changes were found in contralateral kidneys in both p53(+/+) and p53(-/-) mice. Because these chronic changes resemble the age-related pathology and are highly correlated with tubular cell senescence (Chkhotua et al., 2002; Joosten et al., 2003; Chkhotua et al., 2001; Ding et al., 2001), we observed the course of tubular cell senescence in the late phase after IRI.
The positive staining of SA-b-gal has been reported as a characteristic marker for demonstration of cell senescence (Dimri et al., 1995). Positive staining is expressed by various types of cells upon senescence in culture and increases with age in vivo. The activity of SA-b-gal can be histochemically detected at pH 6.0 by X-gal staining (Dimri et al., 1995; Sigal et al., 1999). In the present study, we have found that, in IRI kidneys of p53(+/+) mice, faint staining for SA-b-gal activity appeared at 1 month, but increased significantly at 3 and 6 months after IRI. In contrast, in the contralateral kidneys of p53(+/+) mice and in both the IRI and contralateral kidneys of p53(-/-) mice at 1 and 3 months after IRI, there was almost no positive staining. Positive staining was occasionally detected in IRI kidneys of p53 (-/-) mice at 6 months. This finding also provides in situ evidence that mouse tubular epithelial cells, especially in the corticomedullary portion of the IRI kidneys, were subjected to senescence caused by IRI, and the senescent cells gradually accumulated in vivo after IRI. The senescent cells may initiate early pathological changes in tubulointerstitium. Our results provide direct evidence to support the view that IRI can trigger and accelerate renal cell senescence (Chkhotua et al., 2002; Chkhotua et al., 2005; Joosten et al., 2003; Chkhotua et al., 2001), which is proposed to be one of the mechanisms of the "long-term effect" of IRI.
The involvement of different senescence-inducing pathways maybe depend on the species of cell origin (e.g. human versus rodent) or the type of tissue (BenPorath and Weinberg, 2005; Famulski and Halloran, 2005). Two paradigmatic tumor suppressor proteins, p53 protein and Rb protein, have been confirmed to play critical roles in the induction of senescence. The p53 protein is stabilized and proceeds to activate its transcriptional targets, such as p21 (Kulju and Lehman, 1995). Rb is found at senescence in its active, hypophosphorylated form, which binds to the E2F-protein family members to repress their transcriptional targets (Narita et al., 2003). These targets are the major effectors involved in cell-cycle progression. To investigate the effect of the p53-Rb signaling pathway on tubular cell senescence in the late phase of IRI, we evaluated p53, p21, and Rb expression in p53 (+/+) mouse kidneys, and p21 and Rb expression in p53 (-/-) mouse kidneys. Semi-quantitative immunohistochemistry analysis and quantitative western blot analysis of these proteins demonstrated a marked increase in p53, p21, and hypophosphorylated Rb proteins in IRI kidneys of p53 (+/+) mice at 1, 3, and 6 months after IRI. In p53 (-/-) mice kidneys, however, the expression of p21 and hypophosphorylated Rb were delayed and were much lower than those in p53 (+/+) mice (p<0.05). However, their expression was higher than those in the contralateral kidneys (no IRI kidney). Meanwhile, the expression of SA-b-gal was positively correlated with the expression of each cell cycle regulatory protein in mice of both genotypes (p<0.01). Enhanced expression of these proteins and SA-b-gal were localized in tubular epithelial cells, especially in the corticomedullary portion of the IRI kidneys. Our findings suggest that the p53-Rb signaling pathway may play a major role in tubular cell senescence induced by IRI. The p53 protein can be activated by various types of stress, such as telomere uncapping, DNA damage, oxidative stress, oncogene activation, and others (Hasty and Vijg, 2004; Campisi, 2005b; Price et al., 2004; Supavekin et al., 2003; Cao et al., 2003; Tyner et al., 2002; Maier et al., 2004). In the IRI kidney, oxidative stress and DNA damage are persistent, even in the late phase after IRI (Gueler et al., 2004; Molitoris and Sutton, 2004; Khundmiri et al., 2004). This stress may lead to a continuous activation of p53, which results in the activation of the p53-Rb signaling pathway and, eventually, triggers and maintains tubular cell senescence in the late phase of IRI. On the contrary, p53 deficiency can sufficiently prevent tubular cell senescence induced by IRI. In mouse embryo fibroblasts (MEFs), inactivation of p53 is also sufficient in preventing senescence, allowing these cells to divide indefinitely (Dirac and Bernards, 2003). On the other hand, cells that carry a null mutation in the Rb gene have normal senescence, yet inactivation of additional Rb family members p107 and p130 is sufficient in preventing senescence (Dannerberg et al., 2000; Sage et al., 2000). This data indicates that both p53 and Rb are necessary for the initiation of senescence. Once MEFs have undergone senescence, the continued activity of both p53 and Rb is required to maintain this state, and inactivation of either of these genes in senescent MEFs allows these cells to resume a proliferating state (Dirac and Bernards, 2003). These findings suggest a model of a linear activation pathway, in which a stress signal activates p53, which in turn activates Rb. The p21 protein, an inhibitor of cyclin E/Cdk2 complexes, is the natural candidate to mediate this linear activation of Rb by p53. In our experiment, the expression of p21 protein was distinctly inhibited in the kidneys of p53(-/-) mice, which was notably accompanied by a lack of staining for SA-b-gal activity. This finding indicates that p21 is a crucial link in the p53-Rb signaling pathway in tubular cell senescence caused by IRI. Nevertheless, deletion of p53 can not thoroughly prevent the expression of p21 or Rb and the occurrence of tubular cell senescence. We propose that p21 can be activated directly by a p53-independent pathway, such as DNA damage or telomere erosion (Chkhotua et al., 2005; Joosten et al., 2003; Ding et al., 2001; Ben-Porath and Weinberg, 2005; Megyesi et al., 2001; Price et al., 2004). Rb can also be activated by the p16 protein (Famulski and Halloran, 2005; Devarajan et al., 2003; Megyesi et al., 2001), which is an inhibitor of cyclin D/Cdk4, 6 complexes and is not normally expressed in adult tissues. However, p16 is induced in various situations of stress and is highly expressed in senescent cells. Rb can, thus, be activated either by the p53-p21 pathway, or by the p16 pathway, or by both in parallel. The activation levels of p53 and Rb in different cell types are related to the specific combination of stressors and their severity. Our study showed that the activation of the p53-p21-Rb pathway was the major event in tubular cell senescence, and the activation of the p16-Rb pathway was the secondary event after clamping the left renal hila for 45 min and re-establishing reflow for more than 1 month. Of course, this should be further studied and verified.
In summary, the current study indicates that IRI can accelerate renal tubular cell senescence, which is presumed to be one of the mechanisms of the "long-term effect" of IRI.Tubular cell senescence caused by IRI may play an important role in the initiation and/or progression of tubulointerstitial fibrosis. The activation of the p53-p21-Rb signaling pathway may play a vital role in the onset and progression of tubular cell senescence induced by IRI. Inactivation or down-regulation of p53 activity may be one of the therapy strategies for reducing or partly preventing the "long-term effect" of IRI.
Acknowledgements
This study was supported by a grant from Chung Kong Scholar Programme, Ministry of Education of the People's Republic of China and by the Special Funds for the Major State Basic Research Program of China (973 program). (No. 2005CB522604).
References
1. Azuma H, Nadeau KS, MackenzieH, et al (1997). Nephron mass modulates the hemodynamic, cellular, and molecular response of the rat renal allograft. Transplantation 63: 519-528. [ Links ]
2. Ben-Porath I, Weinberg RA (2005). The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 37: 961-976. [ Links ]
3. Bodnar AG, Ouellette M, Frolkis M, et al (1998). Extension of lifespan by introduction of telomerase into normal human cells. Science. 279: 349-352. [ Links ]
4. Brenner BM, Milford EL (1993). Nephron underdosing: a programmed cause of chronic renal allograft failure. Am J Kidney Dis. 21: 66-72. [ Links ]
5. Campisi J (2005a). Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120: 513-522. [ Links ]
6. Campisi J (2005b). Aging, tumor suppression and cancer: high wireact! Mech Ageing Dev. 126: 51-58. [ Links ]
7. Cao L, Li W, Kim S, et al (2003). Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 17: 201-213. [ Links ]
8. Chen PL, Scully P, Shew JY, et al (1989). Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell 58: 1193-1198. [ Links ]
9. Chkhotua A, Shapira Z, Tovar A, et al (2001). Cellular senescence: a new marker of kidney function recovery after ischemic injury in rats. Transplant Proc. 33: 2910-2915. [ Links ]
10. Chkhotua A, Shohat M, Tobar A, et al (2002). Replicative senescence in organ transplantation-mechanisms and significance. Transpl Immunol. 9: 165-171. [ Links ]
11. Chkhotua AB, Schelzig H, Wiegand P, et al (2005). Influence of ischaemia/reperfusion and LFA-1 inhibition on telomere lengths and CDKI genes in ex vivo haemoperfusion of primate kidneys. Transpl Int. 17: 692-698. [ Links ]
12. Daemen MA, Ven MW van de, Heineman E, Buurman WA (1999). Involvement of endogenous interleukin-10 and tumor necrosis factor-alpha in renal ischemia-reperfusion injury. Transplantation. 67: 792-800. [ Links ]
13. Dannenberg JH, Rossum A van, Schuijff L, Riele H te (2000). Ablation of the retinoblastoma gene family deregulates G (1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 14: 3051-3064. [ Links ]
14. Devarajan P, Mishra J, Supavekin S, et al (2003). Gene expression in early ischemic renal injury: clues towards pathogenesis, biomarker discovery, and novel therapeutics. Mol Genet Metab. 80: 365-376. [ Links ]
15. Dimri GP, Lee X, Basile G, et al (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 92: 9363-9367. [ Links ]
16. Ding G, Franki N, Kapasi AA, et al (2001). Tubular cell senescence and expression of TGF-beta1 and p21(WAF1/CIP1) in tubulointerstitial fibrosis of aging rats. Exp Mol Pathol. 70: 43-53. [ Links ]
17. Dirac AM, Bernards R (2003). Reversal of senescence in mouse fibroblasts through lentiviral suppression of p53. J Bio Chem. 278: 11731-11734. [ Links ]
18. Donehower LA, Harvey M, Slagle BL, et al (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 356: 215-221. [ Links ]
19. Epstein M (1996). Aging and the kidney. J Am Soc Nephrol. 7: 1106-1122. [ Links ]
20. Es A van, Hermans J, Bockel JH van, et al (1983). Graeff J de: Effect of warm ischemia time and HLA (A and B) matching on renal cadaveric graft survival and rejection episodes.Transplantation 36: 255-258. [ Links ]
21. Famulski KS, Halloran PF (2005). Molecular events in kidney ageing. Curr Opin in Nephrol Hypertens. 14: 243-248. [ Links ]
22. Goyal VK (1982). Changes with age in the human kidney. Exp Gerontol. 17: 321-331. [ Links ]
23. Gueler F, Gwinner W, Schwarz A, et al (2004). Long-term effects of acute ischemia and reperfusion injury. Kidney Int. 66: 523-527. [ Links ]
24. Haller H, Dragun D, Miethke A, et al (1996). Antisense oligonucleotides for ICAM-1 attenuate reperfusion injury and renal failure in the rat. Kidney Int. 50: 473-480. [ Links ]
25. Halloran PF, Melk A, Barth C (1999). Rethinking chronic allograft nephropathy: the concept of accelerated senescence. J Am Soc Nephrol. 10: 167-181. [ Links ]
26. Hasty P, Vijg J (2004). Accelerating aging by mouse reverse genetics: a rational approach to understanding longevity.Aging Cell. 3: 55-65. [ Links ]
27. Joosten SA, Ham V van, Nolan CE, et al (2003).Telomere shortening and cellular senescence in a model of chronic renal allograft rejection. Am J Pathol. 162: 1305-1312. [ Links ]
28. Kang DH, Kanellis J, Hugo C, et al (2002). Role of the microvascular endothelium in progressive renal disease. J Am Soc Nephrol. 13: 806-816. [ Links ]
29. Kelly KJ, Plotkin Z, Dagher PC (2001). Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J Clin Invest. 108: 1291-1298. [ Links ]
30. Kelly KJ, Plotkin Z, Vulgamott SL, Dagher PC (2003). P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol. 14: 128-138. [ Links ]
31. Khundmiri SJ, Asghar M, Khan F, et al (2004). Effect of ischemia and reperfusion on enzymes of carbohydrate metabolism in rat kidney. J Nephrol. 17: 377-383. [ Links ]
32. Kulju KS, Lehman JM (1995). Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res. 217: 336-345. [ Links ]
33. Linderman RD, Tobin J, Shock NW (1985). Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc. 33: 278-285. [ Links ]
34. Maier B, Gluba W, Bernier B, et al (2004). Modulation of mammalian life span by the short isoform of p53. Genes Dev. 18: 306-319. [ Links ]
35. McLaren BK, Zhang PL, Herrera GA (2004). P53 protein is a reliable marker in identification of renal tubular injury. Appl Immunohistochem Mol Morphol. 12: 225-229. [ Links ]
36. Meguid El, Nahas A, Bello AK (2005). Chronic kidney disease: the global challenge. The Lancet. 365: 331-340. [ Links ]
37. Megyesi J, Andrade L, Jr Vieira JM, et al (2001). Positive effect of the induction of p21WAFI/CIP1 on the course of ischemic acute renal failure. Kidney Int. 60: 2164-2172. [ Links ]
38. Melk A, Kittikowtt W, Sandhu I, et al (2003). Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 63: 2134-2143. [ Links ]
39. Melk A, Schmidt BM, Takeuchi O, et al (2004). Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 65: 510-520. [ Links ]
40. Molitoris BA, Sutton TA (2004). Endothelial injury and dysfunction: Role in the extension phase of acute renal failure. Kidney Int. 66: 496-499. [ Links ]
41. Narita M, Nunez S, Heard E, et al (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703-716. [ Links ]
42. Nogae S, Miyazaki M, Kobayashi N, et al (1998). Induction of apoptosis in ischemia-reperfusion model of mouse kidney: Possible involvement of Fas. J Am Soc Nephrol. 9: 620-631. [ Links ]
43. Price PM, Megyesi J, Saf Irstein RL (2004). Cell cycle regulation: Repair and regeneration in acute renal failure. Kidney Int. 66: 509-514. [ Links ]
44. Sage J, Mulligan GJ, Attardi LD, et al (2000). Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14: 3037-3050. [ Links ]
45. Schonthal A, Feramisco JR (1993). Inhibition of histone H1 kinase expression, retinoblastoma protein phosphorylation, and cell proliferation by the phosphatase inhibitor okadaic acid. Oncogene 8: 433-441. [ Links ]
46. Shih W, Hines WH, Neilson EG (1988). Effects of cyclosporin A on the development of immune-mediated interstitial nephritis. Kidney Int. 33: 1113-1118. [ Links ]
47. Sigal SH, Rajvanshi PR, Gorla G, et al (1999). Parial hepatectomy-induced polyploidy attenuates hepatocyte replication and activates cell aging events. Am J Physiol. 276: G1260-G1272. [ Links ]
48. Supavekin S, Zhang W, Kucherlapati R, et al (2003). Differential gene expression following early renal ischemia/reperfusion. Kidney Int. 63: 1714-1724. [ Links ]
49. Taylor LM, James A, Schuller CE, et al (2004). Inactivation of p16INK4a, with retention of pRb and p53/p21cip1 function, in human MRC5 fibroblasts that overcome a telomere-independent crisis during immortalization. J Biol Chem. 279: 43634-43645. [ Links ]
50. Tyner SD, Venkatachalam S, Choi J, et al (2002). p53 mutant mice that display early ageing-associated phenotypes. Nature 415: 45-53. [ Links ]
Received on May 22, 2006. Accepted on March 19, 2007.

