










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkBiocell
versión impresa ISSN 0327-9545
Biocell v.32 n.1 Mendoza ene./abr. 2008
A tridimensional view of the organization of actin filaments in the central nervous system by use of fluorescent photooxidation
Francisco Capani1, Ezequiel Saraceno1, Valeria Romina Boti1, Laura Aon-Bertolino1, Juan Carlos Fernández1, Fernando Gato1, Maria Sol Krause2, Lisandro Giraldez3, Mark H. Ellisman4, and Héctor Coirini1,2.
1. Dept. Bioquímica Humana, Facultad de Medicina, Universidad de Buenos Aires, Paraguay 2155 - 5° P, (1121) Buenos Aires, Argentina.
2. Instituto de Biología y Medicina Experimental, Vuelta de Obligado 2490, (1428) Buenos Aires, Argentina.
3. Laboratorio de Neuroquímica e Biología Celular, Instituto de Ciencias da Saúde, Universidad Federal da Bahia, Bahia, Brasil.
4. National Center for Electron Microscopy and Imaging Research, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0608, USA.
Address correspondence to: Francisco Capani, MD, PhD. Departamento de Química Humana, Facultad de Medicina, Universidad de Buenos Aires, Paraguay 2155 - 3ºP, (1121) Buenos Aires, ARGENTINA. E-mail: fcapani@fmed.uba.ar
ABSTRACT: Cellular and subcellular organization and distribution of actin filaments have been studied with various techniques. The use of fluorescence photo-oxidation combined with phalloidin conjugates with eosin has allowed the examination of the precise cellular and subcellular location of F-actin. Correlative fluorescence light microscopy and transmission electron microscopy studies of F-actin distribution are facilitated with this method for morphological and physiological studies. Because phalloidin-eosin is smaller than other markers, this method allows the analysis of the three-dimensional location of F-actin with high-resolution light microscopy, three-d serial sections reconstructions, and electron tomography. The combination of selective staining and three-dimensional reconstructions provide a valuable tool for revealing aspects of the synaptic morphology that are not available when conventional electron microscopy is used. By applying this selective staining technique and three-dimensional imaging, we uncovered the structural organization of actin in the postsynaptic densities in physiological and pathological conditions.
Keywords: F-actin; Phalloidin-eosin; Phootoxidation; 3-D reconstructions; Electron tomography.
Introduction
Actin is a ubiquitous protein that is able to form polar filamentous structures which are the major part of the cytoskeleton eukaryotic cell. Intracellular actin is present in monomer form (G-actin) or polymerized filament form (F-actin). Once it reaches a critical concentration, G-actin (with ATP) forms F-actin and eventually reaches a steady state. In the steady state, ATP is hydrolyzed into ADP, and the length of filament becomes stable (Korn et al., 1987). Observations with electron microscopy (EM) reveal that, at the steady state, F-actin may form massive filament networks (Hartwin, 1992). A number of proteins that regulate actin cytoskeleton have been identified, including cofilin (also named actin depolymerization factor) Arp 2/3, and formins (Bamburg et al., 1999; Chen et al., 2000; Pollard et al., 2000; Wear et al., 2000; Condeelis, 2001; Holt and Koffer, 2001; Winder and Ayscough, 2005; Pollard, 2007). Arp2/3 complex is active at the leading edge of motile cells, where it produces branches on the sides of existing filaments. Formins nucleate and support the elongation of unbranched actin filaments, which cooperate in rapidly assembling profilin-actin into long filaments while remaining continuously associated with the fast-growing barbed end (Kovar, 2006). Formins associate processively with the fast-growing ends of filaments and protect them from capping. In contrast, cofilin can bind to F-actin in the interface between two subunits and effectively depolymerized the filament.
Most of the methods that are currently used to detect actin (Fifkova and Delay, 1982; Landis and Reese, 1983; Matus et al, 1982) can not distinguish between monomeric globular (G) and filamentous (F) actin. In the last decade, phalloidin (a mushroom toxin) conjugated with different fluorophores has been extensively used to study the distribution of actin in cells and tissues (Cooper, 1987; Capani et al., 2001a, b and c; Fukazawa et al., 2003; Ouyang et al., 2005). Phalloidin has been primarily used in studies of F-actin in cultured cells by use of light microscopy (LM). The mushroom toxin phalloidin is a small peptide consisting of seven amino acids with a molecular weight of 789 Kda. Phalloidin binds to both large and small F-actin with high affinity but, unlike antibodies, does not bind to G-actin (Wulf et al., 1979; Cooper, 1987). Although fluorescent molecules are not in themselves visible under the EM, they can be used to drive the oxidation of diaminobenzidine (DAB) to create a reaction product that can be rendered electron dense (Deerinck et al., 1994; Gaietta et al., 2002). This technique is known as fluorescence photo-oxidation (Maranto, 1982; Sandell and Masland, 1988; Deerinck et al., 1994). Although there are many fluorophores, only a few of them are efficient as photo-oxidizers (Deerinck et al., 1994). Eosin possesses moderate fluorescence, is a potent generator of ROS, and is far superior to more conventional fluorophores (Capani et al., 2001a, b and c; Ouyang et al., 2005; Saraceno et al., 2007).
Taking advantage of these characteristics of phalloidin-eosin compound, we used fluorescent photo-oxidation to study F-actin distribution in the central nervous system (CNS) using LM and two- and three-dimensional (3-D) models under Electron Microscopy (EM). We have provided a brief review of the technique developed during the course of these studies.
Selective staining of actin wiht phalloidin-eosin
Photo-oxidation has become a very useful technique for performing correlative fluorescence LM and EM studies. Correlated microscopy allows for a mapping of proteins with LM and higher resolution location at the ultrastructural level with EM. Photoconversion consists of intense illumination of a dye in fixed specimens in the presence of oxygen and DAB. Dye-catalyzed formation of singlet oxygen causes highly localized polymerization of DAB into an insoluble osmiophilic precipitate visible by EM (Fig 1). Using this approach, we have uncovered multiple morphological features in the F-actin-rich structures in the CNS (Capani et al., 2001a, b). Because eosin is a small molecule with good penetration, we were also able to perform 3D analysis with fluorescence LM and EM.

FIGURE 1. Confocal and electron microscopic images of phalloidin labeling in the rat brain. (A) and (B): Phalloidin labeling in the cerebellar cortex using phalloidin-eosin (A). Punctate staining corresponds to the dendritic spines. In addtion to the puntacte staining, we observed strong staining in the pinceau region (PI), the glomeruli (GL), and surrounding the blood vessels (BV). (B) High magnification photograph of the cerebellar molecular layer showing the details of the puntacte staining. (C) Phalloidin labeling in cerebellar cortex. This image was taken directly from the EM embedding tissue subjected to photooxidation before sectioning. The photoxidation spot was very easy to detect then we were able to choose the correct area to study. (D) EM photograph showing that the reaction product was concentrated in spine heads and neck (arrows). AT: axonal terminal. Scale bar (A and B) = 10 μm; C = 30 μm; D = 1 μm.
Actin is highly concentrated in dendritic spines and serves as a major skeletal protein in the subcellular structure (Fifkova and Delay, 1982; Matus et al., 1982). The strongest staining was seen in areas known to be rich in dendritic spines (Fig.1A-B), as reported in studies that used structural and immunocytochemical methods. (Matus et al., 1982; Cáceres et al., 1983).
The LM observations were highly correlated with the EM observations after photo-oxidation; the dendritic spines showed the most remarkable staining (Fig. 1CD). Moreover, this technique gave us new insights into actin-cytoskeleton organization in the spines. By combining fluorescence photo-oxidation with the tannic acid fixation, we have detected bundles of actin filaments between the lamellae of the spine apparatus (Fig. 1EF). At the LM level, we observed a penetration of phalloidin through the entire section. However, at the EM level, the penetration was only 8 um (Capani et al., 2001a, b and c).
Dendritic spines also showed consistent staining when we injected cells with fluorescent phalloidin in fixed tissue (Fig. 2A-B). Although the result of staining the spines was fairly conventional after the introduction of the antibodies and the phalloidin, the selectivity with this injected phalloidin-staining revealed more spines emanating from dendrites than are typically visible with intracellular injection of fluorescent dyes, such as Lucifer Yellow, a result that is consistent with LM studies using tissue and cultured neurons (Capani et al., 2001a, b; Comery et al., 1997; Deerinck et al., 1994). Further measurements are needed to quantify the relevance of this difference.

FIGURE 2. Individual Purkinje Cell neurons injected either with Alexa 568 and phalloidin conjugated to Alexa 488. (A) Projection of a series of optical sections through a Purkinje neuron showing the dendritic tree staining with Phalloidin-Alexa 488. (B and C) Single slices showing a portion of the dendritic tree displaying either the Alexa 568 channel (B) or phalloidin-Alexa 488 channel. Observe that more spines are stained in the cells injected with phalloidin-Alexa 488 than with Alexa 568. Scale bar = 10 μm.
Unfortunately, we were unable to inject eosin-phalloidin with use of the same protocol, most likely because the eosin-phalloidin conjugate may not possess sufficient charge for iontophoresis. Furthermore, when we tried to inject eosin-phalloidin by pressure injection, the eosin-phalloidin got stuck in the micropipette (Capani et al., 2004). Using phalloidin histochemistry and image correlation spectroscopy (ICS), we have quantified dendritic spines (Wiseman et al., 2002). Thus, we think that fluorescent phalloidin might be useful for the visualization and quantification of dendritic spines on individual neurons using LM.
Three dimensional visualization of actin filaments with use of electron tomography and 3-D reconstructions
A disadvantage of the analysis of individual thin sections is the loss of three-dimensional (3D) information about the structure of relatively large subcellular structures. Three-dimensional reconstructions were performed at higher resolution with both serial section reconstruction or electron tomography. Because photooxidation is dependent on ROS release (Capani et al., 2001), the lack of the photo-oxidation product deep in the tissue might be related to the lack of oxygen. Nevertheless, the depth of the labelling was sufficient to study the 3-D distribution of F-actin in the dendritic spines. Using this technique, we found that F-actin was concentrated in only the head of mushroom-type spines in the hippocampus areas CA1, CA3, dentate gyrus, and neostriatum (Fig. 3A-B).

FIGURE 3. Three-dimensional reconstructions of spines stained with phalloidin-eosin followed by photo-oxidation. (A-D) Three-dimensional serial section reconstructions of dendritic spines F-actin positive from neostriatal tissue. F-actin staining was concentrated in the head of the spines (yellow). No F-actin was concentrated either in the presynaptic terminal (red) or in the the dendritc shaft (green). (E) Computed slices through tomographic volumes of hippocampal CA1. Bundles of actin filaments were seen connecting ER to PSD (arrows). (F) Electron tomographic reconstruction of the dendrite spine head. Spines apparatus (blue and yellow) is connected to the post-synaptic density (magenta) through the F-actin filaments. Scales bars: (A, B) = 1 μm; (C,D) = 0.5 μm.
Intermediate voltage electron microscopy (IVEM) is a not a complicated method for obtaining 3-D information about a unique stained structure, such as phalloidin-stained spines. IVEMs operate at higher accelerating voltages (300,000-3,000,000 eV) than conventional transmission electron microscopes (TEMs). The higher accelerating voltages allow for the use of thicker sections than is possible with conventional TEMs. Section thickness is limited to 0.25 mm or less with conventional TEMs, whereas sections up to 10 mm-thick have been examined with the more powerful high-voltage electron microscopes (Woodcock et al., 1991; Bouwer et al., 2007). In sections of this thickness, a relatively large population of synapses can be visualized in their total number and size. A recent advance in the field of IVEM is the application of electron tomography to the analysis of structures contained within these thicker sections. Electron tomography, such as in the more familiar medical tomographic techniques, uses a series of projections taken at different angles of an object to create a 3-D volume reconstruction. In the case of electron tomography, the specimen is tilted around a fixed axis and images are acquired at regular tilt increments over a wide tilt range, usually 75º (for overview, see Koster et al., 1997; Lucic et al., 2005). Images are aligned and a volume reconstruction of the specimen is created by back projection (Fig. 2). Electron tomography of thick sections has several advantages for 3-D analysis compared to serial section or stereo pair analysis. In addition to the ease of preparing a single thick section compared to a series of consecutive thin sections, alignment errors due to missing sections or section distortions are avoided because no physical sectioning is involved. This feature is of tremendous benefit when trying to follow tortuous, extended structures with fine features, such as mitochondrial cristae (Perkins et al., 1997), th Golgi apparatus (Ladinsky et al., 1994, 1999) and astrocytes (Bushong et al., 2001). Unlike stereo pairs, tomographic volumes also allow the object to be rotated and resectioned along any axis, thereby revealing internal structure and providing easy correlation of 2-D and 3-D images of the same structure (see Fig. 3). In recent years, tomographic analysis has been used with great success to study and obtain quantitative information on the 3-D structure of synapses and related structures such as spiny dendrites (Martone et al., 1999) and specialized synaptic complexes in frogs and chicks (Lenzi et al., 1999; Shoop et al., 1999).
Electron tomography also has the advantage of producing computed slices that are thinner than can be prepared using physical sectioning (Martone et al., 1999). Using tomography, we were able to show the continuity of the actin bundles between the lamellae of the spine apparatus (SA) with the post-synaptic density (PSD), a feature hardly seen in single EM sections. (Fig. 3C and D). Thus, fluorescence photo-oxidation combined with 3D reconstruction techniques allowed us to study the F-actin location in different populations of spines, which provided additional details about the organization of the actin cytoskeleton in these structures.
Although staining was most concentrated in the spines, we also observed intense staining in a subset of additional structures in the CNS, including the cerebellar glomerulli, certain axons and dendrites, some astrocytic processes, and the pinceau area in the cerebellum (Capani et al., 2001c). The pinceau area showed very strong and consistent staining. In a stereopair from a thick section, we found the strongest labelling in basket-cell axons (Fig. 6), which was correlated with the strong labelling observed by confocal microscopy (Fig. 1). Thus, phalloidin-eosin photo-oxidation offers the possibility for gaining additional information about Factin rich structures in CNS at high resolution by use of EM analysis.
Recently, quantum dots have been introduced to perform pre-embedding multi-protein labelled for correlative light and electron microscopy studies. Although the penetration and the preservation was good with the use of detergents, some of them are detectable only by use of magnification of at least 25,000 x (Giepmans et al., 2005). Furthermore, recombinant proteins containing tetracysteine tags have been successively labeled in living cells with different colors of biarsenical fluorophores so that older and younger protein molecules can be sharply distinguished by both fluorescence and electron microscopy. In this case, photooxidation was used to study the trafficking of connexin only in vitro with reasonably good resolution (Gaietta et al., 2002; Hoffman et al., 2005). However, in this study, they are not able to perform a correlative in vivo study in contrast with the unique possibility that offers photooxidation with phalloidin-eosin to work in vivo.
Significance of the F-actin distribution
The application of photoconversion phalloidinesoin staining and imaging techniques can reveal new aspects of brain function in structures that are highly
rich in actin. By using this powerful combination, we examined the change in F-actin in dendritic spines in hippocampal slices in more detail by using EM. Labeling of F-actin was observed in many dendritic spines in a non-tetanized region of a hippocampal slice. In contrast, this F-actin labeling in dendritic spines was decreased by approximately 50% after 30 min in the LTP area of the same slice (Ouyang et al., 2005). Therefore, induction of LTP is associated with an initial depolymerization of F-actin in dendritic spines that may be related with the transport of molecules from and to the synaptic terminal (Ouyang et al., 2005). Consistent with this data, fluorescence photo-oxidation was used by Fukazawa et al. in 2003 to study the function of F-actin in synaptic plasticity after LTP stimulation. Induction of LTP has been reported to induce an early increase the F-actin content of dendritic spines in hippocampus in vitro and in vivo (Fukazawa et al., 2003; Okamoto et al., 2004) and to modify the size of the spines (Chen et al., 2007). Finally, F-actin cytoskeleton is involved in the organization and dynamic of presynaptic endocytic process (Shupliakov et al., 2002; Capani et al., 2004).
Recently, using the same technique, we observed an increment of F-actin staining of the neostriatal dendritic spines of 6-month-old birth-hypoxic animals (Fig. 4A-D). Under hypothermia conditions, these changes were diminished dramatically (Saraceno et al., 2007). This data is consistent with previous studies performed on hippocampal tissue (Gisselsson et al., 2005).
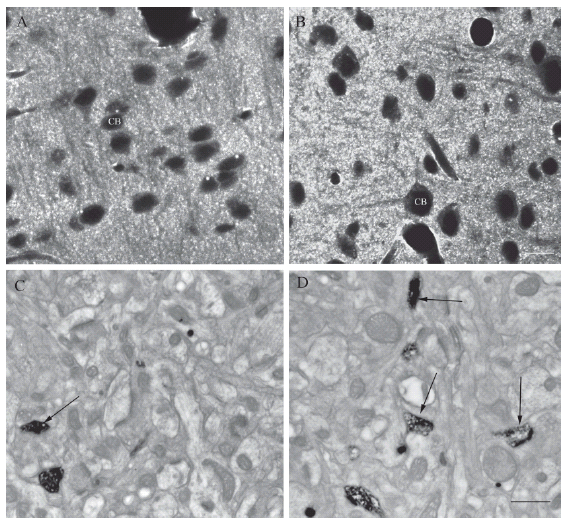
FIGURE 4. Correlative confocal and photooxidation electron microscopic pictures in neoxcortex of control (A and C) and rats sujected to hypoxia (B and D). Note the increment in the fluorescence intensity between A and B. EM microphotographs obtained from photooxidazed tissue show and increment in the number of dendritic spines (arrows) in hypoxia conditions. Scale bar, A and B = 10 μm; C, D = 1 μm.
Conclusions
We have demonstrated that the application of fluorescence photo-oxidation plus phalloidin eosin can reveal new aspects of brain F-actin structures. This technique also possesses several advantages over traditional methods for locating F-actin. Mainly among these advantages was the possibility to use high concentration of glutaraldehyde (up to 1%) in the primary fixative, which resulted in a greatly improved ultrastructural preservation compared to inmunocytochemical methods. We were also able to obtain a good spatial distribution when we used 3-D reconstruction techniques. However, we were not able to detect F-actin in areas where the concentration was very low or the actin cytoskeleton suffered dynamic and rapid changes in its organization, as occurs in the presynatic terminal. Finally, phalloidin photo-oxidation may be useful to study functional changes in the actin network.
Acknowledgements
This work was supported by ANPCyT BID 1728/OC-AR PICT 15001, UBACYT M020.
References
1. Bamburg JR, McGough A, Ono S (1999). Putting a new twist on actin: ADF / cofilins modulate actin dynamics. Trends Cell Biol. 9: 364-370. [ Links ]
2. Bouwer JC, Mackey MR, Lawrence A, Deerinck TJ, Jones YZ, Terada M, Martone ME, Peltier ST, Ellisman MH (2007). The application of energy-filtered electron microscopy to tomography of thick, selectively stained biological samples. Methods Cell Biol. 79: 643-660. [ Links ]
3. Bushong EA, Martone ME, Jones YZ, Ellisman MH (2001). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 22: 183-192. [ Links ]
4. Caceres A, Payne MR, Binder LI, Steward O (1983). Immunocytochemical localization of actin and microtubule-associated protein MAP2 in dendritic spines. Proc Natl Acad Sci U S A. 80: 1738-1742. [ Links ]
5. Capani F, Martone ME, Deerinck TJ, Ellisman MH (2001a). Selective localization of high concentrations of F-actin in subpopulations of dendritic spines in rat central nervous system: a three-dimensional electron microscopic study. J Comp Neurol. 435: 156-170. [ Links ]
6. Capani F, Deerinck TJ, Ellisman MH, Bushong E, Bobik M, Martone ME (2001b). Phalloidin-eosin followed by photo-oxidation: a novel method for localizing F-actin at the light and electron microscopic levels. J Histochem Cytochem. 49: 1351-1361. [ Links ]
7. Capani F, Ellisman MH, Martone ME (2001c). Filamentous actin is concentrated in specific subpopulations of neuronal and glial structures in rat central nervous system. Brain Res. 923: 1-11. [ Links ]
8. Capani F, Andersson F, Crum J, Lillig C, Tomilin N, Evergren E, Ellisman MH, Brodin L, Shupliakov O (2004). Directionality of the actin filament assembly in the synaptic endocytic zone. Program No. 967.1. Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience. [ Links ]
9. Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A. 94: 5401-5404. [ Links ]
10 Condeelis J (2001). How is actin polymerization nucleated in vivo? Trends Cell Biol. 11: 288-293. [ Links ]
11. Cooper JA (1987). Effects of cytochalasin and phalloidin on actin. J Cell Biol. 105: 1473-1478. [ Links ]
12. Chen H, Bernstein BW, Bamburg JR (2000). Regulating actin-filament dynamics in vivo. Trends Biochem Sci. 25: 19-23. [ Links ]
13. Chen LY, Rex CS, Casale MS, Gall CM, Lynch G (2007). Changes in synaptic morphology accompany actin signaling during LTP. J Neurosci 27: 5363-5372. [ Links ]
14. Deerinck TJ, Martone ME, Lev-Ram V, Green DP, Tsien RY, Spector DL, Huang S, Ellisman MH (1994). Fluorescence photooxidation with eosin: a method for high resolution immunolocalization and in situ hybridization detection for light and electron microscopy. J Cell Biol. 126: 901-910. [ Links ]
15. Fifkova E, Delay RJ (1982). Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol. 95: 345-350. [ Links ]
16. Fukazawa Y, Saitoh Y, Ozawa F, Ohta Y, Mizuno K, Inokuchi K (2003). Hippocampal LTP is accompanied by enhanced Factin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38: 447-460. [ Links ]
17. Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296: 503-507. [ Links ]
18. Giepmans BN, Deerinck TJ, Smarr BL, Jones YZ, Ellisman MH (2005). Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat Methods. 10: 743-748. [ Links ]
19. Gisselsson LL, Matus A, Wieloch T (2005). Actin redistribution underlies the sparing effect of mild hypothermia on dendritic spine morphology after in vitro ischemia. J Cereb Blood Flow Metab. 25: 1346-1355. [ Links ]
20. Hartwin L (1992). An ultrastructural approach to understanding the cytoskeleton. In: The Cytoskeleton - a Practical Approach. Carraway, K.L. & Carraway, C.A.C. (Eds), Oxford University Press, Oxford, pp. 23-45. [ Links ]
21. Holt MR, Koffer A (2001). Cell motility: proline-rich proteins promote protrusions. Trends Cell Biol. 11: 38-46. [ Links ]
22. Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Ellisman MH, Lohse MJA (2005). FlAsH-based FRET approach to determine G proteincoupled receptor activation in living cells. Nat Methods 3: 171-176. [ Links ]
23. Korn ED, Carlier MF, Pantaloni D (1987). Actin polymerization and ATP hydrolysis. Science. 238: 638-644. [ Links ]
24. Koster AJ, Grimm R, Typke D, Hegerl R, Stoschek A, Walz J, Baumeister W (1997). Perspectives of molecular and cellular electron tomography. J Struct Biol. 120: 276-308. [ Links ]
25. Kovar DR (2006). Molecular details of formin-mediated actin assembly. Curr Opin Cell Biol. 18: 11-17. [ Links ]
26. Landis DMD, Reese TS J (1983). Cytoplasmic organization in cerebellar dendritic spines. J Cell Biol 97: 1169-1178. [ Links ]
27. Ladinsky MS, Kremer JR, Furcinitti PS, McIntosh JR, Howell KE (1994). HVEM tomography of the trans-Golgi network: structural insights and identification of a lace-like vesicle coat. J Cell Biol. 127: 29-38. [ Links ]
28. Ladinsky MS, Mastronarde DN, McIntosh JR, Howell KE, Staehelin LA (1999). Golgi structure in three dimensions: functional insights from the normal rat kidney cell. J Cell Biol. 144: 1135-1149. [ Links ]
29. Lenzi D, Runyeon JW, Crum J, Ellisman MH, Roberts WM (1999). Synaptic vesicle populations in saccular hair cells reconstructed by electron tomography. J Neurosci. 19: 119-132. [ Links ]
30. Lucic V, Forster F, Baumeister W (2005). Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem. 74: 833-865. [ Links ]
31. Maranto AR (1982). Neuronal mapping: a photooxidation reaction makes Lucifer yellow useful for electron microscopy. Science 217: 953-955. [ Links ]
32. Martone ME, Jones YZ, Young SJ, Ellisman MH, Zivin JA, Hu BR (1999). Modification of Postsynaptic density after transient cerebral ischemia: A quantitative and Three-dimensional ultrastructural study. J Neurosci. 15: 1988-1997. [ Links ]
33. Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K (1982). High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sc. USA. 79: 7590-7594. [ Links ]
34. Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 7: 1104-1112. [ Links ]
35. Ouyang Y, Wong M, Capani F, Rensing N, Lee CS, Liu Q, Neusch C, Martone ME, Wu JY, Yamada K, Ellisman MH, Choi DW (2005). Transient decrease in F-actin may be necessary for translocation of proteins into dendritic spines. Eur J Neurosci. 12: 2995-3005. [ Links ]
36. Perkins GA, Renken CW, Young SJ, Lamount SP, Martone ME, Lindsey S, Frey TG, Ellisman MH (1997). Electron Microscopy of large multicomponent biological structures. J Struct Biol. 120: 219-227. [ Links ]
37. Pollard TD, Blanchoin L, Mullins RD (2000). Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 29: 545-576. [ Links ]
38. Pollard TD (2007). Regulation of actin filament assembly by arp2/3 complex and formins. Annu Rev Biophys Biomol Struct. 36: 451-477. [ Links ]
39. Sandell JH, Masland RH (1988). Photoconversion of some fluorescent markers to a diaminobenzidine product. J Histochem Cytochem. 36: 555-559. [ Links ]
40. Saraceno E, Aon L, Boti V, Kruse M, Fernández JC, Valverde D, Gato F, Madureira D, Giraldez Alvarez L, Coirini H, Capani F (2007). Hypoxia induces actin cytoskeletal changes in neostriatal dendritic spines. Program No. 869.1.Abstract Viewer/Itinerary Planner. Washington, DC: Society for Neuroscience. [ Links ]
41. Shupliakov O, Bloom O, Gustafsson JS, Kjaerulff O, Low P, Tomilin N, Pieribone VA, Greengard P, Brodin L (2002). Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci USA. 99: 14476-14481. [ Links ]
42. Wear MA, Schafer DA, Cooper JA (2000). Actin dynamics: assembly and disassembly of actin networks. Curr Biol. 10: R891-R895. [ Links ]
43. Woodcock CL, McEwen BF, Frank J (1991). Ultrastructure of chromatin. II. Three-dimensional reconstruction of isolated fibers. J Cell Sci. 99: 107-114. [ Links ]
44. Winder SJ, Ayscough KR (2005). Actin binding proteins. J Cell Sci. 118: 651-654. [ Links ]
45. Wiseman PW, Capani F, Squier JA, Martone ME (2002). Counting dendritic spines in brain tissue slices by image correlation spectroscopy analysis. J Microsc. 205: 177-186. [ Links ]
46. Wulf E, Deboben A, Bautz A, Faulstich H, Wieland TH (1979). Fluorescent phallotoxin, a tool for the visualization of cellular actin. Proc Natl Acad Sci USA 76: 4498-4502. [ Links ]
Received on August 6, 2007.
Accepted on September 17, 2007.

