










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Asociación Química Argentina
Print version ISSN 0365-0375
An. Asoc. Quím. Argent. vol.92 no.1-3 Buenos Aires Jan./July 2004
REGULAR PAPERS
Electrochemical Reduction Of Carbon Dioxide In The Presence Of [NiII–5,7,12,14-Tetramethyldinaphtho [b,i] [1,4,8,11] Tetra Aza[14]Annulene]++ Cation.
A. Rios-Escudero1, M. Isaacs1, M. Villagrán1, J. Zagal1 and J. Costamagna1.
1Faculty of Chemistry and Biology, Universidad de Santiago de Chile
Av. B. OHiggins 3363, P.O. Box 40, Santiago 33 Chile.
FAX: (56-2) 681-2108, Email: jcostama@lauca.usach.cl
Received January 06, 2004. In final form March 19, 2004
Dedicated to Professor Dr. P. J. Aymonino on occasion of his 75thAnniversary
Abstract
Cyclic Voltammograms of [Ni(II)5,7,12,14-tetrametyldinaphtho[b,i] [1,4,8,11]tetraaza[14]annulene]++ cation complex ([Ni(tmdnTAA)]2+ hereafter) present two irreversible reduction peaks at -0.70 V y -1.50 V vs. Ag/AgCl in N,N´-dimethylformamide (DMF), while in acetonitrile (MeCN) these peaks are at -0.93V. and -1.80 V vs. Ag/AgCl. When DMF/H2O or MeCN/H2O are used as solvents, the peaks are also shifted with respect to those in pure DMF as solvent. For example, in DMF/ H2O they are -0.76 and -1.04 V respectively. Cyclic voltammograms of the cation complex in CO2 saturated solutions exhibited the catalytic wave of the CO2 reduction, Ecp, at ~ -1.50 V in DMF/H2O and at ~ -1.80 V in MeCN/H2O. Under these conditions, the only product of the bulk electrolysis was hydrogen. Due to the formation of a stable product, the catalytic current for the CO2 reduction of the second voltammetric cycle is 30% less intense than the current of the first cycle. The stable adduct, [Ni(II)tmdnTAA-CO]++, formed on the electrode was characterizated by spectroeletrochemistry and FT-IR spectroscopy.
Resumen
Voltamogramas cíclicos del catión complejo macrocíclico [Ni(II)-1,7,12,14-tetrametildinafto[b,i] [1,4,8,11] tetraaza[14]anuleno]++, ([Ni(II) tmdnTAA]++), presenta dos picos de reducción irreversibles a -0.70 V y -1.50 V vs. Ag/AgCl en solución de N,N dimetilformamida (DMF), mientras que en solución de acetonitrilo(MeCN) los potenciales de pico se desplazan a -0.93 V y -1.80 V respectivamente. La voltametría cíclica del complejo en una solución saturada con CO2 muestra una onda catalítica de reducción del CO2 cercana a -1.50V para la mezcla DMF/H2O y a -1.80V para la mezcla de MeCN/H2O. En todos lo experimentos de electrólisis a potencial controlado solamente fue detectado hidrógeno como producto. Además, la corriente de pico catalítica para la reducción de CO2 se reduce en un 30% al segundo ciclo voltamétrico lo cual indica el envenenamiento de la superficie del electrodo por formación de un aducto estable del tipo [Ni(II)tmdnTAA-CO]++. Este aducto fue caracterizado por espectroelectroquímica y medidas de FT-IR.
Introduction
There has been considerable interest in the design of electrocatalysts for carbon dioxide reduction to useful fuel products [1-5]. A number of transition metal complexes in both homogeneous solutions or confined on electrode surfaces have been shown to be effective in the electrocatalytic reduction of carbon dioxide [1-13]. The electroreduction of CO2 involves not only multiple electron transfers but also complex chemical steps which may involve protonation [1,3,14,15,16]. Furthermore, there can be multiple and competing reaction pathways giving rise to a variety of reaction products and some of them can deactivate the catalyst. In the mechanism of electrode poisoning when the reduction of CO2 is catalyzed by [Ni(CRH)]2+ (CR= 2,12- dimethyl-3,7,11,17-tetraazabicyclo-[11,3,1]heptadeca-1(17),2,11,13,15-pentaene) or [Ni(cyclam)]2+ (cyclam= 1,4,8,11-tetraazacyclotetradecane), the CO formed reacts with Ni(I) to form a carbonyl adduct. The adsorbed Ni(I) carbonyl is further reduced to the catalytically inactive Ni(0) carbonyl [14]. On the basis of these precedents, we have investigated the electroreduction of CO2 on glassy carbon electrodes in the presence of [Ni(II)-5,7,12,14-tetramethyldinaphtho[b,i][1,4,8,11]tetraaza[14]annulene]2+, [Ni(II)(tmdnTAA)]++, shown in Figure 1.

Figure 1: -Molecular structure for: [Ni(II)-5,7,12,14-tetramethyldinaphtho[b,i][1,4,8,11]tetraaza[14]annulene]++, [Ni(tmdnTAA)]++.
Experimental
The synthesis, spectroscopic characterization, acid-base properties, and photochemistry behavior of [Ni(tmdnTAA)]++, and [Cu(tmdnTAA)]++ macrocyclic cation complexes have been previously reported [17]. Electrochemical measurements were carried out with a PAR-173 potentiostat coupled to a PAR-175 universal programmer, and a X-Y Omnigraphic recorder. All potentials are referred to the Ag/AgCl electrode without correction for the liquid junction. Deaerated solutions 10-4 M in [Ni(tmdnTAA)]Cl2 and 0.01M in tetraethylammoniumn perclorate (TEAP), were placed in a three electrode cell with a glassy carbon working electrode (GC), platinum wire counter electrode and a Ag/AgCl reference electrode. All cyclic voltammetry experiments were performed at 200 mV/s.
Electrolysis experiments in saturated CO2 atmosphere were carried out in the same configuration used for cyclic voltammetry. A three electrode airtight cell with a Duocel reticulated glassy carbon(RVC) 10PPI working electrode was used in these experiments. A homemade thin layer cell similar to one described by C.M.Duff and G.A.Heath [18] was used. An optical transparent Pt mesh electrode was the working electrode, a platinum wire was the counter electrode, and Ag/AgCl was the reference electrode. This thin layer cell configuration was placed inside a Varian Cary 1E spectrometer. In a typical spectro-electrochemical experiment, the spectra were recorded at three minutes intervals until constant absorbance. In the CO saturated atmosphere experiment the same configuration was used. Reaction products were analyzed by a Varian 3400 Gas Chromatograph equipped with TCD and FID double detectors, respectively. The capillar column used with the TCD detector was Molesiv 115-3632, 30m length, and 0.53 mm I.D. The capillary column used with the FID was a DB-1.
The products formaldehyde and formic acid were investigated using the chromotropic acid spot test [19]. A chromatographic column of 30 cm x 1.5 cm was packed with silica gel, and then the electrolysis products were separated using a 1:1 (v\v) chloroform-methanol mixture as eluent.
FT-IR spectra for bulk electrolysis products were measured in a Bruker model Vector 22 apparatus using a film sample over a KBr window.
Results And Discussion
Voltammetric behavior of [Ni(tmdnTAA)]++ cation.
In general, the complex investigated was electrochemically well behaved and exhibited both metal-based as well as ligand-based irreversible redox processes under nitrogen, carbon dioxide and carbon monoxide atmospheres. Under nitrogen, the first electron reduces the metal center [20] to generate the [Ni(I)(tmdnTAA)]+ species. This process is followed by the addition of another electron to generate the ligand-reduced

Figure 2.- Voltammograms under nitrogen(¾¾) and carbon dioxide(- - - -) atmospheres for the macrocycle studied in N,N-dimethyl formamide/tetraethylammonium perchlorate (TEAP) at v=200 mV/s.
Table I. Redox potentials for [Ni(II)-5,7,12,14-tertramethyl-dinaphtho [b,i][1,4,8,11]tetraaza[14] annuleneH]++ at a glassy carbon electrode in 0,01M tetraethylammonium perchlorate, Under N2 and CO2 atmosphere.
| Solvents | Metal-based(a) | Ligand-based(a) | Ered (b ) |
| DMF | -0.70 | -1.50 | -1.75 |
| MeCN | -0.93 | - 1.80 | - 1.80 |
| DMF/H2O | -0.76 | -1.04 | -1.50 |
| MeCN/H2O | -1.10 | -1.80 | -1.80 |
(a) Potential in Volts, vs. Ag/AgCl, in N2.
(b)Potentials in Volts, vs. Ag/AgCl, in CO2. Ered values represents the potential of the wave at which the current enhancement due to electrocatalytic reduction of carbon dioxide was observed for the macrocycle studied.
species [Ni(I)(tmdnTAA)]. Typical voltammograms for these processes are shown in Figure 2. Table 1 contains peak potential values for the irreversible processes under nitrogen and carbon dioxide atmospheres in N,N-dimethylformamide (DMF), Acetonitrile (MeCN), DMF/H2O, and MeCN/ H2O solvents.
The last column of Table I lists the potentials at the peak of the wave at which the current enhancement due to electrocatalytic reduction of carbon dioxide was observed in the solvents studied. The electrocatalytic activity of the Ni(II) macrocycle complex toward reduction of carbon dioxide was evaluated by comparing its voltammetric response under nitrogen and carbon dioxide atmospheres as it can be seen in Fig. 2. A clear electrocatalytic current was detected in the presence of the Ni(II) macrocycle complex. The reduction of carbon dioxide observed at –1.8V (vs. Ag/AgCl) in the absence of the complex takes place at a potential more negative than –2.0 V(11). On the other hand, [Ni(tmdnTAA)]++ has always shown electrocatalytic current after the second reduction process as it is shown in Figure 2. Cyclic voltammograms in protic media exhibit an anodic feature at –0.05 V when the limiting potential was more negative than the potential where the hydrogen evolution began, Figure 3. We propose that the feature corresponds to the oxidation of a nickel hydride formed on the electrode surface, eqns (1) and (2).
| [Ni(II)(tmdnTAA)]+++e-→[Ni(I)(tmdnTAA))]+ads | (1) |
| [Ni(I)(tmdnTAA)]+ads + H+ →[HNi(II)(tmdnTAA)]++ ads | (2) |
Similar electrochemical processes were reported by Aramata et al. for the reduction of carbon dioxide on a glassy carbon electrode chemically modified by aza-macrocyclic-cobalt(II) complexes [21,22].
In our eletrochemical experiments with the Ni(II) complex in protic media, an enhancement of the carbon dioxide electrocatalytic current was evidenced in the voltammograms and also hydrogen evolution occurred in DMF and MeCN wet solvents. Furthermore, the catalytic current of the second cycle, e.g., in DMF/ H2O, is ~30% less than the current of the first cycle. At the same time, shifts to a more positive catalytic potential were observed in DMF/H2O respect to dry DMF solvent (see Table 1), and the hydrogen evolution is also increased. These results suggest a possible competition between eq. (5) and eq. (6).
| [Ni(II)(tmdnTAA)]+++ e-+ CO2→ [N(I)(tmdnTAA) CO2]+ | (3) |
| [Ni(I)(tmdnTAA)CO2]+ + H →[Ni(I)(tmdnTAA) COOH ]++ | (4) |
| [Ni(I)(tmdnTAA)COOH]+++ e-→ [Ni(II)(tmdnTAA)CO]+++OH- | (5) |
| [HNi(II)(tmdnTAA)]++ads+H++e-→[Ni(II)(tmdnTAA)]++ads+H2 | (6) |

Figure 3.- Voltammogram of [Ni(tmdnTAA)]++ in N,N-dimethylformamide after addition of water (showing the anodic hump at –0.05 V), under nitrogen (¾¾) and carbon dioxide (× × × ×) at v=200 mV/s.
The reaction mechanism proposed by Sauvage et al. [15] and Anson et al. [16] for the electrocatalytic reduction of carbon dioxide with [NiCyclam]++ over a mercury electrode can be applied to the reduction catalyzed with [Ni(tmdnTAA)]++ . In case of the latter, the nickel(II) carbonylated complex shown in equation (5) is very stable, and this may explain the low efficiency of the electrocatalytic reduction with [Ni(tmdnTAA)]++ cation complex.
In MeCN solutions only the second cathodic peak electrocatalyzed the carbon dioxide reduction at –1.80V (see Table I). If protons were added to the media, e.g. as a wet solvent, the voltammogram of N2-saturated solutions exhibited the anodic hump of the hydride oxidation also observed in wet DMF solutions (see Figure 3). A very strong enhancement of the electrocatalytic peak and the absence of the hydride-related anodic hump resulted in wet MeCN solutions saturated with carbon dioxide. This experimental observations suggest that in acetonitrile and in protic media the reaction corresponding to equation 6 would not take place (at ca. –1.80V).
Preparative–scale electrolysis.
Preparative–scale electrolysis was performed in a gas-tight electrolysis cell. The current decayed slowly and the solution turned from red to yellow over a period of three hours under an applied potential of –1.80V vs. Ag/AgCl. Samples of the gas phase were collected at thirty minute intervals. Hydrogen was the only reaction product as shown in figure 4.

Figure 4.-Time-evolution of hydrogen for a bulk electrolysis experiment for [Ni(tmdnTAA)]++ under carbon dioxide in N,N-dimethylformamide /water(1:1) solvent at -1.65V vs. Ag/AgCl.
Analytical tests for oxalate, formic acid, and formaldehyde in the liquid phase were negative for all the cases studied. Spectroelectrochemical observations were made in the presence of carbon dioxide or carbon monoxide at open circuit and -1.8 V, they revealed a new absorption band at 358 nm as shown in figure 5. The absorption band can be attributed to a stable species [Ni(tmdnTAA)CO]++. A similar but unstable species was detected by Anson et al. in the electrocatalytic reduction of carbon dioxide to carbon monoxide by [Ni(cyclam)]2+ [16]. Similar experiments in a carbon monoxide atmosphere have shown the same behavior as presented in Figure 6 (see experimental).
The FT-IR spectra of electrolyzed and non electrolyzed [Ni(tmdnTAA)]++ solutions were recorded after chromathography on silica gel. The results are shown in figure 7 for an electrolysis of a solution saturated with nitrogen, for an electrolysis of a solution saturated with carbon dioxide, and non electrolyzed [Ni(tmdnTAA) ]++ cation. The spectra from the non electrolyzed and the electrolysis under nitrogen are similar (Fig. 7a, 7b) but the spectrum from the electrolysis under carbon dioxide exhibited a new band at 1729 cm-1

Figure 5.- Spectroelectrochemistry for [Ni(tmdnTAA)]++ in acetonitrile under carbon dioxide saturated atmosphere at open circuit (O.C.) and for Eap.(applied potential)= -1.8 V. See inset for details.
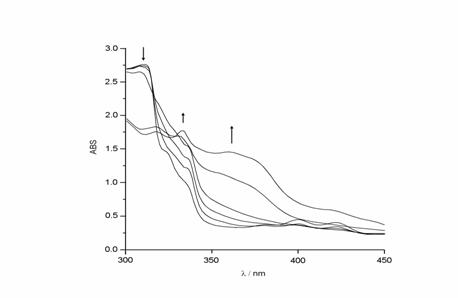
Figure 6.- Spectroelectrochemistry for [Ni(II) (tmdnTAA)]++ at Eap = -1.8 V in acetonitrile, under carbon monoxide saturated atmosphere, showing the spectral changes each 3 minutes until constant behavior.
corresponding to the carbonyl [Ni(tmdnTAA)-CO]++ complex. Therefore, this stable adduct, detected by spectroelectrochemical and FT-IR spectroscopy, could be responsible for the catalyst poisoning when carbon dioxide is electroreduced in DMF, DMF/H2O, MeCN or MeCN/H2O solutions.

Figure 7.- FT-IR film spectra of: a) [Ni(tmdnTAA)]++; and the electrolysis products: b) under nitrogen, c) under carbon dioxide.
Conclusions
The fact that catalytic currents were observed after the second reduction process, suggest that probably two electrons are involved in the carbon dioxide electroreduction. In addition, it is quite likely that more than one mechanism can explain the behaviour observed in cyclic voltametry.
It is also clear that the nature of the products and their distribution depend on the amount of water present in the system for the macrocycle studied.
The [Ni(II)-5,7,12,14-tetramethyldinaphtho[b,i][1,4,8,11]tetraaza[14] annulene]++ cation complex showed electrocatalytic effect in cyclic voltammetric experiments for carbon dioxide reduction over GC electrode in the media studied. Electrolysis experiments at –1.8V vs. Ag/Ag/Cl showed the formation of a stable adduct [Ni(II)(tmdnTAA)CO]++ which would be responsible of the loss of catalytic actvity, and the absence of chromathographic detection of carbon monoxide product in the bulk electrolysis experiments. These results have shown that the stable Ni(II) carbonyl complex is the catalytically inactive species in the system studied. The selectivity and efficiency for the hydrogen evolution in the catalytic process can be modified as a function of the amount of water added.
Acknowledgements
This work has been financed by FONDECYT (Chile), project Nº 8010006,M.I. thanks FONDECYT (Chile), project Nº 2000010. A.R.E. thanks FONDECYT (Chile) for a doctoral fellowship.
References
[1] Costamagna, J.; Ferraudi, G.; Canales, J.; Vargas, Coord. Chem. Rev. 1996, 148, 221. [ Links ]
[2] Costamagna, J.; Ferraudi, G.; Matsuhiro, B.; Campos-Vallete, M.; Canales, J.; Villagrán, M.; Vargas, J.; Aguirre, M.J., Coord. Chem. Rev. 2000, 196, 125. [ Links ]
[3] Keene, F.R.; Sullivan, B.P., Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, Sullivan, B.P.; Krist, K.; Guard, H.E., Eds., Elsevier: Amsterdam, 1993; Chap. 5. [ Links ]
[4] Bhugun, I.; Lexa, D. ; Saveant, J-M. ; J. Am. Chem. Soc. 1996, 118, 1769. [ Links ]
[5] Bhugun, I.;Lexa, D.; Saveant, J-M. ; J. Phys. Chem. 1996, 100, 1981. [ Links ]
[6] Mostafa Hossain, A.G.M.; Nagaoka, T.; Ogura, K.; Electrochimica Acta 1997, 42, 2577; [ Links ] Chaplin, R.S.P.; Wragg, A.A.; J. Appl. Electrochem., 2003, 33,1107. [ Links ]
[7] Rasmussen, S.C.; Richter, M.M.; Yi, E.; Place, H.; Brewer, K.J. Inorg. Chem., 1990, 29, 3926. [ Links ]
[8] Noemi, G.; Nallas, A.; Brewer, K.J. Inorg. Chim. Acta, 1996, 253, 7. [ Links ]
[9] Smith, C.I.; Crayston, J.A.; Hay; R.W. J. Chem. Soc., Dalton Trans., 1993, 3267. [ Links ]
[10] Fujita, E.; Half, J.; Sanzenbachen, R.; Elias, H. Inorg. Chem., 1994,33, 4627. [ Links ]
[11] Beley, M.; Collin, J.P.; Rupert, R.; Sauvage, J.P. J. Chem. Soc., Chem. Commun., 1984, 1315. [ Links ]
[12] Leiber, C.M.; Lewis, N.S. J. Am. Chem. Soc., 1984, 106, 5033. [ Links ]
[13] Fujihia, M.; Hirata, Y.; Suga, K. J. Electroanal, Chem., 1990, 292, 199. [ Links ]
[14] Bujno, K.; Bilewicz, R.; Siegfried, L.; Kaden, T. Electrochim. Acta, 1997, 42, 1206. [ Links ]
[15] Beley, M; Collin, J.P.; Ruppert, R.; Sauvage, J.P. J. Am. Chem. Soc., 1986, 108, 7461. [ Links ]
[16] Balazs, G.B.; Anson, F.C. J. Electroanal. Chem., 1993, 361, 149. [ Links ]
[17] Costamagna, J.; Ferraudi, G.; Villagran, M.; Wolcan, E. J. Chem. Soc., Dalton Trans., 2000, 2631, [ Links ]
[18] Duff, C.M.; Heath, G.A. Inorg. Chem., 1991, 30, 2528. [ Links ]
[19] Arana, C.; Yan, S.; Keshavarz, M.; Potts, K.T.; Abruña, H.D. Inorg. Chem., 1992, 31, 3680. [ Links ]
[20] a) A. Rios-Escudero, M. Villagrán, J. Costamagna, G. Ferraudi., J. Coord. Chem., 2003, 56, 1233. [ Links ] b) A. Rios-Escudero, J. Costamagna, G. I. Cardenas-Jiron., J. Phys. Chem. A., 2003, submitted for publication. [ Links ]
[21] Tanaka, H.; Aramata, A. J. Electroanal. Chem., 1997, 431, 29. [ Links ]
[22] Aga, A.; Aramata, A.; Hisaeda, Y. J. Electroanal. Chem., 1997, 437, 111. [ Links ]

