



![Electrochemical reduction of carbon dioxide in the presence of [NiII-5,7,12,14-tetramethyldinaphtho [b,i] [1,4,8,11] tetra aza [14]annulene]++ cation](/img/es/prev.gif)






Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Asociación Química Argentina
versión impresa ISSN 0365-0375
An. Asoc. Quím. Argent. v.92 n.1-3 Buenos Aires ene./jul. 2004
REGULAR PAPERS
A Model For The Dissolution Of Metal Oxides Mediated By Heterogeneous Charge Transfer
García Rodenas1,2, L.A.; Araujo1,2, P.; Bruyère1,3, V.I.E.; Morando1,3, P.J.; Regazzoni1,3, A.E.; Blesa1,2, M.A.
1Unidad de Actividad Química, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica (CNEA), Avenida General Paz 1499, 1650, San Martín, Provincia de Buenos Aires
Phone: + (54 11) 6772-7161; Fax: + (54 11) 6772-7886
e-mail: miblesa@cnea.gov.ar
2 Escuela de Posgrado, Universidad Nacional de San Martín (UNSAM)
3 Instituto de Tecnología J. Sabato (UNSAM-CNEA)
ReceivedFebruary 18, 2004. In final form March 19, 2004
Dedicated to Prof. Pedro J. Aymonino on the occasion of his 75th birthday
Abstract
The experimental characterization of charge transfer transitions (LM and IV CTs) in surface complexes formed on iron oxides and TiO2, together with the measurements of the rate of magnetite dissolution in the presence of pentacyanoisonicotinatoferrate(II) are used to build and validate a general model for the dissolution of metal oxides mediated by charge transfer processes. A critical surface ensemble, denoted ºS, is identified for the different cases. For reductive dissolution of iron(III) oxides, ºS is often an iron(II) surface complex that dissolves in a first order kinetic process; the steady state concentration of ºS, and hence its rate of dissolution is determined by the balance between its rate of formation and either the rate of oxo-bond breakage or the rate of diffusion/scavenging of the conjugate oxidised form of the reductant. In the case of IVCT within dimeric surface complexes, we postulate that the vibrational activation required for charge transfer also produces labilization; thus, charge transfer and dissolution are concerted processes. Charge trapping by the surface complex following the photochemical formation of an electron-hole pair in the semiconductor also generates a critical ensemble. In this case, however, successful dissolution requires of adequately high rates of removal of both charge carriers, and dissolution is arrested if there are no scavengers for the other charge carrier (the hole, in the case of reductive dissolution). This effect is realized experimentally in the photochemical dissolution of nickel ferrite, with and without oxalic acid as hole scavenger.
Resumen
Se presenta la caracterización experimental de las transiciones de transferencia de carga (LM y IV CT) en complejos superficiales formados sobre óxidos de hierro y TiO2, junto con mediciones de la velocidad de disolución de magnetita en presencia de pentacianoisonicotinatoferrato(II). Los resultados se usan para construir y validar un modelo general de disolución de óxidos metálicos, mediada por procesos de transferencia de carga. Para cada uno de los diferentes casos, se identifica un ensamble superficial crítico, llamado ºS. En la disolución reductiva de óxidos de hierro(III), ºS es frecuentemente un complejo superficial de Fe(II) que se disuelve en un proceso cinético de primer orden; el balance entre su velocidad de formación y ya sea la velocidad de ruptura de uniones oxo o la velocidad de difusión/secuestro de la especie oxidada conjugada determinan la concentración de estado estacionario de ºS y por lo tanto su velocidad de disolución. En el caso de IVCT en complejos diméricos, postulamos que la activación vibracional requerida para la transferencia de carga también produce labilización, de forma que la transferencia de carga y la disolución son procesos concertados. El entrampamiento en complejos superficiales de los electrones y huecos generados en el semiconductor por absorción de luz también produce ensambles críticos. En este caso, sin embargo, la disolución requiere que las velocidades de remoción de los dos portadores de carga sean adecuadamente altas, y la disolución se frena si no hay secuestrador del otro portador de carga (el hueco, en el caso de la disolución reductiva). Este efecto se observa experimentalmente en la disolución fotoquímica de la ferrita de níquel, con y sin ácido oxálico como secuestrador de huecos.
Introduction
Metal oxide dissolution in aqueous media is of importance in a wide variety of fields and applications, including metal corrosion and passivation, weathering of oxide minerals, mobilization of metallic species in the environment and in biological fluids, hydrometalurgy, synthesis of materials by soft methods, etc. Many different mechanisms may be involved, including attack by acids or bases, ligand-assisted dissolution, and charge-transfer mediated processes. The latter process is of importance in the metabolism of dissimilatory iron-reducing bacteria (DIRB) in a specific process that leads either to total dissolution of Fe(III) oxides [1-3], or to the formation of biogenic magnetite; the characteristics of the partially reduced solid thus formed have been used to suggest that the magnetite found in the Martian meteorite Allan Hills 84001 was produced by the action of DIRB [4]. For a description of the state of the knowledge until 1993, see reference [5] and references therein.
The present paper focuses on charge transfer mediated dissolution. Oxides prone to redox dissolution include iron(III), manganese(III,IV), chromium(III), uranium(IV), and several other semiconducting metal oxides. In the general case, the systems of interest include the metal oxide, a reductant or oxidant, and complexing agents. Activation by light absorption is also important. Here, we present a spectroscopic characterization of the charge transfer bands in surface complexes (formed upon adsorption) that either mediate dissolution reactions or illustrate the important features of the transitions. We further present measurements of the rates of the reductive dissolution of magnetite by pentacyanoferrate(II) complexes, Fe(CN)5Ln- (L = pyridine, isonicotinamide and isonicotinate), and of the rates of the photo-assisted dissolution of nickel ferrite with and without oxalic acid. The results, together with the information available in the literature, are used to postulate a model of the coupling of heterogeneous charge transfer with dissolution.
Experimental
The oxides used in this study were lepidocrocite (d-FeOOH), hematite (a-Fe2O3), magnetite (Fe3O4), nickel ferrite (NiFe2O4), and TiO2. Except for TiO2, which was a commercial sample from Degussa (P-25) containing ca 80% anatase and 20% rutile, all other oxides were synthesized, as described previously [6-8]. Na4[Fe(CN)5isonic].6H2O was synthesized by reaction of isonicotinic acid with Na3[Fe(CN)5NH3].3H2O in water [9]. Other prussides were synthesized analogously. All other reagents were of analytical grade; water was bidistilled in a quartz apparatus.
UV-visible spectra in solution were recorded on a Shimadzu UV-210A instrument. Diffuse reflectance spectra of the species formed on the surfaces of d-FeOOH and TiO2 upon equilibration with thiocyanate and catechol solutions, respectively, were recorded on the vacuum-dried solids, which were filtered off through 0.2 mm pore size membranes.
FTIR spectra were recorded using a NICOLET 560 instrument equipped with a liquid N2 cooled MCT‑A detector, using a horizontal ZnSe-ATR unit. The incidence angle was 45° and the total number of reflections was 11. Layers of TiO2 particles were deposited by placing TiO2 suspensions (10-20 g dm-3) on the surface of the ATR crystal and evaporating to dryness at room temperature. The excess of TiO2, in the form of loosely adhered particles, was wiped off by a gentle rinsing with water. The coated crystal was mounted in the ATR cell, and allowed to equilibrate with a ligand-free solution at a certain pH and ionic strength until the FTIR signal became stable, and a blank single-beam spectrum (I0) was collected. Then, the concentration of the adsorbate was fixed at a desired value, and the IR-absorption spectra (log(I0/I)) were recorded at 6-10 min intervals until signal amplitudes reached stable values (less than 2% change in 10 min). Bands due to dissolved ligands were negligible within the explored concentration range.
Dissolution experiments were performed at fixed pH values, at 30.0 ± 0.1 oC. The pH was kept constant in the course of the experiment by adding acid from a pH-stat. Ionic strength (m) was controlled through the addition of NaClO4. A typical experiment was started by pouring the oxide onto the thermostated acid solution, or by acidification of a suspension of pH @ 7 (dissolution is negligible at neutral pH). Aliquots were sequentially withdrawn, quenched by cooling and/or alkalization, and filtered through 0.45 mm pore size cellulose acetate membranes. Standard analytical procedures were used to determine the concentration of iron in the filtrates. Fe(II) and total Fe were determined photometrically in the presence of phenanthroline [10]; in the latter case, hydroxylamine was added prior to the determinations. In addition, total Fe was determined photometrically, at 530 nm in thioglicollic acid [11].
Thermal (dark) dissolution of nickel ferrite, NiFe2O4 was performed as described in ref [12]. For the photochemical study, a xenon lamp with an interference filter (I0 = 0.94 ´ 10-5 einstein dm-3 s-1; lmax = 384 nm) was used to irradiate the oxide suspensions. In a typical experiment, dissolution reaction was initiated by adding 20 mg of nickel ferrite to 100 cm3 of 0.1 mol dm-3 oxalate solution at pH 3.5 and m = 0.5 mol dm-3 (NaClO4), previously thermostated at 70 ºC. N2 was bubbled through the solution and the suspension was magnetically stirred throughout the experiment. Aliquots were sequentially withdrawn, filtered, and Fe and Ni were measured spectrophotometrically by the thioglycollic acid method and atomic absorption respectively.
Results
Spectroscopic measurements
Figure 1, taken from [6] and from [13], shows the visible reflectance spectra of the surface complexes formed upon chemisorption of thiocyanate onto d-FeOOH, and of catecholate onto TiO2. LMCT bands of ºFeIII-SCN- and ºTiIV‑cathecolate are centered at 533 and 420 nm, respectively.

Figure 1: Diffuse reflectance spectra of ºFeIII-SCN (a) and ºTiIV-catecholate (b) surface complexes
Albeit slightly red-shifted, they correspond excellently with those of the analogous aqueous complexes. The IR spectrum of the ºTiIV-cathecolate complexes is shown in Figure 2.
The band at 1486 cm-1 is assigned to C-C normal modes of the aromatic ring; the band at 1263 cm-1 corresponds to the stretching of deprotonated, metal-coordinated C-O groups [14].
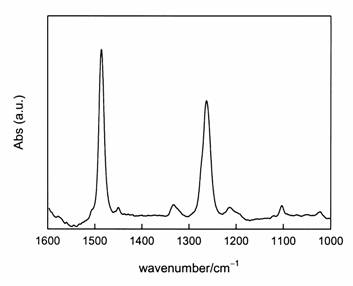
Figure 2: FTIR-ATR spectrum of catechol chemisorbed onto a TiO2 film.
Figure 3 shows the spectrum of the dimer formed in solution by the interaction of FeII(CN)5(isonic)4- (isonic = isonicotinate) with Fe(III).

Figure 3: Visible absorption spectrum of FeIII-O(O)C-C5H4N-FeII(CN)5- at pH 3.7; [Fe(III)] = 2.1 ´ 10-4 mol dm-3; [FeII(CN)5isonic4-] = 8.2 ´ 10-4 mol dm-3.
Other prussides behave similarly but are not shown; here we focus in the isonicotinate complex because it is the most effective to bring about magnetite dissolution (see below). In addition to the typical MLCT transition, located at 407 nm, a broad intervalence (metal to metal) charge transfer (IVCT) band is seen at 740 nm.

Figure 4: Dissolved Fe(III) from magnetite at 30 oC as a function of time at pH 3.30, in the presence of the following concentrations of FeII(CN)5isonic4-: (n) 0.01; (o) 7.03 ´ 10-3; (u) 3.86 ´ 10-3; (m) 2.92 ´ 10-3; (l) 0.001 mol dm-3.
Kinetic measurements
Dissolution profiles of magnetite at pH 3.3 and various FeII(CN)5(isonic)4- concentrations are shown in Figure 4. They are typical of surface-controlled dissolution processes. At pH 3.3 and low adsorbate concentrations, the rate increases linearly with [FeII(CN)5(isonic)4-]; a saturation effect is observed at the highest concentration (Figure 5). Although fitting to a Langmuir equation is not warranted by the data, the adsorption pre-equilibrium can be written as in equation (1); the derived apparent equilibrium constant (in terms of the total FeII concentration) is in the order of 30-40 mol-1 dm3. This value is similar to those found in other systems [15, 16].
| ºFeIII-OH + FeII(CN)4(CNH)(isonicH)2- = ºFeIII-(isonic)-FeII(CN)4(CNH)2- + H2O | (1) |
Although the adsorption affinity is expectedly large for all anions of the type
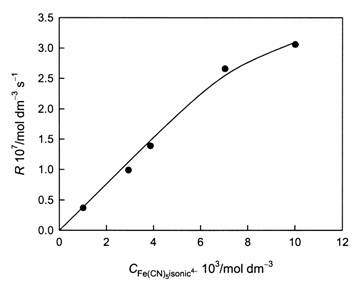
Figure 5: Dependence of the linear dissolution rates on [Fe(CN)5isonic4-]; pH 3.30.
FeII(CN)5Ln-, including FeII(CN)64-, the rates (not shown) of magnetite dissolution in the presence of the related complexes FeII(CN)5(py)3- and FeII(CN)5(ina)3- (py = pyridine, ina = isonicotinamide) are much lower. We conclude that the surface complex that mediates dissolution contains an isonicotinate bridge between surface ºFeIII and adsorbed FeII, as shown in Figure 6.

Figure 6: Surface mixed valence dimeric complex.
In agreement, the IVCT band observed in the spectra of the filtered solutions (Figure 3) demonstrates the presence of {FeII(CN)5(isonic)-FeIII}-. The ability of FeII(CN)5(isonic)4- to interact with the surface through the terminal carboxylate of isonicotinate renders a more favorable adsorption pre-equilibrium, hence faster dissolution.
Figure 7 shows the influence of pH at [FeII(CN)5(isonic)4-] = 4.0 ´ 10-3 mol dm-3. Again, the profiles indicate surface-controlled dissolution, and reflect the effect of pH on the adsorption pre-equilibrium (see below).

Figure 7: Dissolved Fe(III) from magnetite at 30 oC as a function of time in the presence of [Fe(CN)5isonic4-]TOT = 4.0 ´ 10-3 mol dm-3, at various pH values: (n) 1.95; (o) 2.50; (l) 3.00; (D) 3.30; (m) 3.95.
Photochemical experiments
Figure 8 shows the dissolution profiles (dissolved fraction fFe as a function of time) of NiFe2O4 in 0.1 mol dm-3 oxalic acid, pH 3.5 and 70ºC, in the dark and under illumination. Under our experimental conditions, nickel ferrite photodissolves completely in ca. 2 h; thermal dissolution accounts for less than 10% in the same time. In the absence of oxalate, photodissolution is negligible, as shown by the lowest trace in Figure 8. The profiles for Ni (not shown) are identical, indicating congruent dissolution.

Figure 8: Dissolved Fe fraction from NiFe2O4 as a function of time at pH 3.5, m = 0.5 mol dm-3 (NaClO4) and 70ºC: (a) [Ox]TOT = 0.20 mol dm-3 under light; (b) [Ox]TOT = 0.20 mol dm-3 in the dark; (c) [Ox] = 0 under light.
Discussion
The model
The basic tenets of the dissolution model are:
(i) The chemical reactions taking place at the interface can be described using the surface complexation approach [5]. This approach assumes that a surface metal ion and the ensemble of ligands surrounding it can be described as a true chemical entity (the surface complex), largely decoupled from the rest of the solid lattice (Figures 1 and 2), and characterized by well defined thermodynamic and kinetic properties. All interfacial chemical reactions are therefore described by mass-law equations, with defined stoichiometries and equilibrium constants.
(ii) The rate of dissolution is slow enough to guarantee that mass transport is not rate limiting (Figures 4, 5, 7 and 8). Hence, protolytic, complexation, and charge transfer reactions are equilibrated during steady state dissolution.
(iii) The rate of dissolution is controlled by the transfer to the solution of the more labile species of the metal ion. Thus, the oxides of iron(III) dissolve through Fe(II), those of manganese(III,IV) through Mn(II), chromium(III) through Cr(VI), those of uranium(IV) through U(VI), etc. The dependence of the lability on the oxidation state is dictated by the usual properties of the metal ions.
(iv) The aqueous medium is highly unsaturated, and the reverse reaction, the deposition of metal oxide from the solution, can be ignored.
(v) Dissolution is an electrochemical process that involves phase transfer of the two ions that constitute the solid: the metal ion and the oxide ion. At the steady state, the rate of transfer of both ions must be equal and, in principle, the transfer of either ion may be rate limiting. We shall assume that oxide ions are transferred as water molecules, either free, or coordinated to the metal ion; the rate limiting step is the transfer of the metal ion with its coordination sphere. This rate limiting step can be written as a simple phase transfer of a critical surface ensemble, labeled here as ºS; the symbol º represents the linkage of the surface species to the solid framework.
The suite of chemical reactions required to describe the adsorption pre-equilibria that mediate dissolution are:
| ºM-OH + H+ = ºM-OH2+ | (2) |
| ºM-OH = ºM-O- + H+ | (3) |
| ºM-OH + HL = ºM-L + H2O | (4) |
| ºM-OH + HL-M = ºM-L-M + H2O | (5) |
In these equations, HL represents the acid of a complexing anion L-, and HL-M represents a dissolved complex of Mn+ with a ligand with pendant coordinative groups able to link the surface M ion. Equations (2) and (3) represent protolytic equilibria describing the amphoteric properties of the surface, and equations (4) and (5) represent the formation of mononuclear and binuclear surface complexes, respectively.
These pre-equilibria define the surface excess of the critical ensemble, {ºS}, and the observed dissolution rate takes the form of equation (6).
| kuni | (6) |
The dissolution model must describe at least three cases, the operation of all of which have been documented in the literature [5]:
(i) Charge transfer from the ligand to the metal ion (LMCT), involved in many important cases of reductive dissolution of iron(III) oxides.
(ii) Metal to metal charge transfer (IVCT) in bridged binuclear surface complexes. The surface ion may be reduced (reductive dissolution), or oxidised (oxidative dissolution) by exchange with the metal center of the adsorbed complex. This mechanism prevails in many cases, including the oxidative dissolution of chromium(III) oxides, and the auto-accelerated dissolution of iron(III) oxides.
(iii) Photochemically induced formation of the reactive species upon excitation of the oxide electronic structure.
Characteristic times of the involved processes
The information available about the protolytic reactions (2) and (3) indicates that they take place typically in the millisecond-microsecond range [17]. It is not clear if these reactions are limited by diffusion or by the breakage of the bonds.
Surface complexation reactions are also fast, albeit slower than protonation-deprotonation [18]; adsorption of complexing ligands becomes equilibrated within a few minutes [13].
Next, we shall examine the dynamics of charge transfer in the three cases outlined above, assuming that it takes place within ºM-L or ºM-L-M. We shall use M = Fe(III) as an example, M being a +2 reducing metal ion (Fe2+, V2+, Cr2+). The charge transfer steps, following the formation of ºM-L and ºM-L-M, are described by equations (7) and (8), respectively.
| ºFeIII-L- ® ºFeII-L· | (7) |
| ºFeIII-L-MII ® ºFeII-L-MIII | (8) |
Although we shall focus on simple reducing ligands L, and inorganic reductants like low valence metal ions, the reaction is extremely general and biological species may participate and even compete with simple reductants [19]. Reductive dissolution may also be achieved electrochemically, and the advent of atomic force microscopy has permitted recently a very detailed characterization of the sites on which reactions (2)-(8) take place [20].
These processes are in fact transitions to electronically excited states that can be characterized spectroscopically (see Figures 1 and 2). A well documented case is the reductive dissolution of manganese(III,IV) oxides [21, 22]; when catechol is the reductant, electron transfer is fast as compared to the previous complexation equilibria [23]. Whereas the rate of thermal electron transfer (7) may be severely limited by thermodynamics, the rate of the IVCT process is fast, and can be derived using the usual formalism of Hush (equation 9).
| kET @ 1012 exp [-(DGº+l)2/4lkBT] » 2 ´ 103 s-1 | (9) |
In equation (9), DGº is the standard Gibbs energy of the reaction that may be calculated from the redox potential for the couple FeIII(CN)5isonic3-/FeII(CN)5isonic4- (ca. 0.50 V, see [24]) and from the position of the conduction band edge of magnetite (ca. 0.20 V at pH 3, see [25]). l is the reorganization energy, and kB the Boltzmann constant. The reorganization energy can be calculated from the position of the maximum of the IVCT band (Figure 3):
| hnmax = DGº + l | (10) |
In both cases, the rate of the reverse process is even faster. The binuclear surface complex (Figure 6) may be considered a class II complex, thus it is also possible to calculate the extent of the electron delocalization in both metal centers from the position of the maximum nmax, the half-height width of the band Dn1/2, the maximum absorptivity emax, and the distance r between the two metallic centers, using Hushs formalism [26]:
| x2 = (2.05 ´ 10-2)2 (emax Dn1/2/nmax r2)1/2 | (11) |
Using the value r = 510 pm, which is similar to those reported for related complexes [27, 28], the electron delocalization factor is calculated to be x2 = 0.04. This implies a low probability of finding the electron in the nominally FeIII center, a result that can be extended safely to the analogous surface complexes.
The vibrational requirements associated with a similar heterogeneous electron transfer reaction (i.e., Fe(CN)64- onto TiO2 colloidal particles) were characterized by Blackbourn et al. [29]. It was concluded that one particular form of precursor ºTi-OH...(CN)6Fe4- was involved, and that the vibration associated with the bridging cyanide makes one of the most important contributions to the activation barrier; the substitution of isonicotinate for the bridging cyanide provides an inner-sphere pathway, facilitating electron transfer.
In the case of photochemical reactions, the original generation of a pair (ecb, hvb) takes place within 1 fs [30]; subsequent trapping in surface complexes takes place in the range nano- to pico-second; charge transfer with adsorbed species must be fast enough to compete with recombination, within tenths of ms. The dynamics of charge transfer have been studied using mostly non-dissolving oxides, like TiO2, but these figures apply also to well documented cases of redox photodissolution. We shall focus in the reductive dissolution of Fe(III) oxides, but the oxidative dissolution of Cr(III) oxides has also been studied [31].
Comparison with reported dissolution rate constants, and with those derived from Figures 4 and 7, indicates that all processes (2)-(5) are fast as compared to dissolution. Likewise, reactions (7) and (8) are faster, as well as the reverse reactions (thermal deactivation), which are fast enough to compete efficiently with phase transfer (equation 6) (see however below).
The nature of the critical ensemble and the rate limiting step
(i) Irreversible charge injection into the metal center
Strong reductants may inject an electron irreversibly by either outer- or inner-sphere mechanisms; many examples are available in the literature (ascorbate [32], mercaptocarboxylates [16], etc.). If this charge injection follows or precedes a fast proton transfer, there is no limitation associated with surface charge buildup. For example, equation (12) describes the generation of reduced species without change in the surface potential or charge.
| ºFeIII-OH + L- + H+ ® ºFeII-OH2 + L· | (12) |
In this case, ºS can be loosely described as ºFeII-OH2. Documented cases suggest that the initial rates are constant in the first few percents of dissolution [23, 32], and that ºFeII-OH2 reaches a steady state given by (12) and (6). In principle, the rate could saturate at high reductant concentration, due to accumulation of ºFeII-OH2. Saturation effects observed on [L-] are however associated with the adsorption pre-equilibrium (equation 4); note that all surface species comply with the constraint imposed by the surface mass balance. The apparent affinity constants derived from dissolution rates are appreciably lower than those derived from standard measurements of adsorption isotherms. This difference indicates that ºS is not the main species formed by adsorption, but rather some activated species, able to transfer the metal ion from the surface to the solution. Most probably, protonation of oxo bridges is required before breakage can take place.
(ii) Fast reversible charge injection into the metal center
In many cases, including L- = SCN- [6], interfacial charge transfer is equilibrated (equation 13), and is followed by the slow diffusion away of L· (equation 14). Hence, effective reduction of ºFeIII takes place through an unfavorable pre-equilibrium, and depends on the efficient separation of the reaction products, ºFeII-OH2 and L·. The rate of ºFeII-OH2 phase transfer (6), limited by its steady state surface concentration, turns out to be given by equation (15).
| ºFeIII-OH + L- (ads) + H+ | (13) | |
|
| (14) | |
|
| (15) | |
Adsorbed radicals can also be scavenged by other adsorbed species (equation 16), a reaction that is usually followed by the fast desorption or decomposition of the adduct. This case is exemplified by the dissolution of hematite in thiocyanate and iodide solutions. The steady state concentration of the critical ensemble ºS, hence the rate law, becomes a complex function of [L-]; first and second kinetic orders have been diagnosed for SCN- [6] and I- [33], respectively, and synergism, with change in the kinetic order, has been observed for mixtures [33].
| L· (ads) + L- (ads) ® L2·- (ads) | (16) |
A very important, different, possibility arises if the vibrational activation required for charge transfer gives also rise to enhanced dissolution probability. Vibrational activation, in the case of IVCT (e.g., ºFeIII-O2CC5H5N‑FeII(CN)5), leads to coordination environments around both metal centers that permits the radiationless transfer of the electron from one metal ion to the other. As mentioned before, activation involves certain vibrations of the bridging ligand, but it may also involve vibrations associated with the oxo bonds that bridge ºFe to the rest of the solid. Prior protonation pre-equilibria may also facilitate the achievement of the adequate vibrational activation. The data in Figures 4 and 7 can be interpreted in terms of the adsorption of the diprotonated species FeII(CN)4(CNH)(isonicH)2- (see equation 1). Indeed, Figure 9 shows that, at constant complex total concentration, the pH dependence of the rate (derived from the linear part of the dissolution profiles) parallels the buildup of the dianion.
This result is reasonable because it limits the accumulation of surface charge; from results in many systems, it is also obvious that chemisorption is dissociative, the sites of proton adsorption being probably oxide ions.
In such a case, electron transfer may become concerted with phase transfer, and the rate of dissolution is limited by the requirements to reach the adequate vibrational configuration of the dimeric surface complex. We suggest that the observed dissolution of magnetite in the presence of Fe(CN)5(isonic)4- reflects the formation of vibrationally excited ºFeIII-O2CC5H5N-FeII(CN)5, the critical ensemble, ºS, in equation (6). The species released to solution reverts rapidly to its most stable electronic configuration.

Figure 9: Solution speciation of pentacyanoisonicotinateferrate(II): (a) Fe(CN)4(HCN)(isonicH)2-; (b) Fe(CN)5(isonicH)3-; (c) Fe(CN)5(isonic)4-. Dots: experimental dissolution rates.
(iii) Photochemical activation of the metal oxide
Most of the metal oxides of interest are wide bandgap semiconductors. Irradiation with photons of energy larger than the bandgap creates pairs of valence band holes and conduction band electrons. The availability of minory carriers (electrons or holes, depending on the p or n nature of the semiconductor) increases substantially upon irradiation. Surface band bending determines the migration of one of the charge carriers towards the surface, where it may become trapped. Taking iron(III) oxides as an example, the surface traps for electrons may be viewed as the critical ensemble ºS for dissolution. These species do not differ much from those created by LMCT. In this case, however, the excess hole must be removed to prevent enhanced recombination probability (cf. with the diffusion or scavenging of the conjugate oxidant formed in the LMCT).
Previous work demonstrated that the thermal dissolution of NiFe2O4 proceeds at a rate that is intermediate between those of NiO (bunsenite, a slow dissolving oxide) and a-Fe2O3. Reductants accelerate the dissolution of the oxide by providing a faster pathway for Fe(III) transfer [12], and the disruption of the lattice enhances also the phase transfer of Ni(II).[1] Photochemical dissolution rates of iron(III) oxides are also highly sensitive to the presence of reductants, as shown in Figure 8. In the steady state, the rates of removal of electron and holes must be equal, and hence the rate limiting step may become either the phase transfer of ºS or the rate of hole scavenging by oxalate. The stoichiometry of dissolution, ignoring complexation of the dissolved ions, is given in equation (17).
| NiFe2IIIO4(s) + H2C2O4 + 6H+ ® Ni2+ + 2Fe2+ + 2CO2 + 4H2O | (17) |
The critical ensemble, ºFeII–OxH, is in this case generated by the trapping of photoelectrons:
| ºFeIII–Ox + ecb- + H+ ® ºFeII–OxH | (18) |
Conclusions
The phase transfer of an aliovalent metal ion, created by reduction or oxidation of the constituent ion, is involved in the rate limiting step of charge-transfer mediated dissolution of metal oxides. The steady state concentration of these species is controlled by the balance between different reactions, depending on the nature of the metal oxide and of the reductant (oxidant). When charge transfer takes place as an IV transition in a bridged binuclear surface complex, activation of the transition and of the oxo-bond breakage may involve the same vibrational modes, and charge transfer becomes simultaneous with dissolution.
Acknowledgments
Work supported by ANPCyT (PICT 06-06631), European INCO Project ICA4-2002-1000, and CNEA (P5-PID-36-1 and P5-PID-36-4). PJM, AER and MAB are members of CONICET.
Notas
[1] Oxidants also enhance the dissolution by generating labile Ni(III) (unpublished results). This aspect is not studied in the present paper
References
[1] Zachara, J.M.; Frederickson, J.K.; Li, S.M.; Kennedy, D.W.; Smith, S.C.; Gassman, P.L. Am. Mineral. 1998, 83, 1426 [ Links ]
[2] Kostka, J.E.; Wu, J.; Nealson, K.H.; Stucki, J.W. Geochim. Cosmochim. Acta 1999, 63, 3705 [ Links ]
[3] Sokolov, I.; Smith, D.S.; Henderson, G.S.; Gorby, Y.A.; Ferris, F.G. Environ. Sci. Tecnol. 2001, 35, 341 [ Links ]
[4] Thomas-Keprta, K.L.; Clemett, S.J.; Bazylinski, D.A.; Kirschvinik, J.L.; McKay, D.S.; Wentworth, S.J.; Vali, H.; Gibson, E.K.; Romanek, C.S. Appl. Environ. Microbiol. 2002, 68, 3663 [ Links ]
[5] Blesa, M.A.; Morando, P.J.; Regazzoni, A.E. Chemical Dissolution of Metal Oxides, CRC Press, Boca Raton, Fla. 1994 [ Links ]
[6] Regazzoni, A.E.; Blesa, M.A. Langmuir1991, 7, 473 [ Links ]
[7] Regazzoni, A.E.; Urrutia, G.A.; Blesa, M.A.; Maroto, A.J.G. J. Inorg. Nucl. Chem. 1981, 43, 1489 [ Links ]
[8] Regazzoni A.E.; Matijevic E. Corrosion 1982, 38, 212 [ Links ]
[9] Morando, P.J.; Bruyère, V.I.E.; Blesa, M.A. Trans. Met. Chem. 1983, 8,99 [ Links ]
[10] Vogel A.I. Química Analítica Cuantitativa, Kapelusz 1961, Vol. 2, p. 915 [ Links ]
[11] Leussing, D.L.; Newman, L. J. Amer. Chem. Soc. 1956, 78, 552 [ Links ]
[12] Figueroa, C.A.; Sileo, E.E.; Morando, P.J.; Blesa, M.A. J. Colloid Interface Sci.2000, 225, 403 [ Links ]
[13] Rodríguez, R.; Blesa, M.A.; Regazzoni, A.E. J. Colloid Interface Sci.1996, 177, 122 [ Links ]
[14] Martin, S.T.; Kesselman, J.M.; Park, D.S.; Lewis, N.S.; Hoffmann, M.R. Environ. Sci. Technol. 1996, 30, 2535 [ Links ]
[15] Ballesteros, M.C.; Rueda, E.H.; Blesa, M.A. J. Colloid Interface Sci. 1998, 201, 13 [ Links ]
[16] Borghi, E.B.; Morando, P.J.; Blesa M.A. Langmuir 1991, 7, 1652 [ Links ]
[17] Astumian, R.D.; Sasaki, M.; Yasunaga, T.; Schelly, Z.A. J. Phys. Chem. 1981, 85, 3832 [ Links ]
[18] Grossl, P.R.; Eick, M.; Sparks, D.L.; Goldberg, S.; Ainsworth, C.C. Environ. Sci. Technol. 1997, 31, 321 [ Links ]
[19] Royer, R.A.; Dempsey, B.A.; Jeon, B.H.; Burgos, W.D. Environ. Sci. Technol. 2004, 38, 187 [ Links ]
[20] Hasse, U.; Nieben, J.; Scholz, F. J. Electroanal. Chem. 2003, 556, 13 [ Links ]
[21] Banerjee, D.; Nesbitt, H.W. Am. Mineral. 2000, 85, 817 [ Links ]
[22] Stone, A.T. Environ. Sci. Technol. 1987, 21, 979. [ Links ]
[23] Matocha, C.J.; Sparks, D.L.; Amonette, J.E.; Kukkadapu, R.K. Soil Sci. Soc. Amer. J. 2001, 65, 58 [ Links ]
[24] Toma, H.E.; Malin, J. Inorg. Chem. 1973, 12, 1039 [ Links ]
[25] Vago, E. Estudio de la Reacción de Electroreducción de Oxígeno sobre Electrodos de Óxidos de Hierro Bien Caracterizados. Ph. D. Thesis, 1993, Universidad de Buenos Aires [ Links ]
[26] Creutz, C. Progress Inorg. Chem. 1983, 30, 1 [ Links ]
[27] Fluck, E.; Inoue, H.; Nagao, M.; Yangisawa, S. J. Inorg. Nucl. Chem. 1979 , 41, 287 [ Links ]
[28] Toma, H.E. J. Inorg. Nucl. Chem. 1976, 38, 431 [ Links ]
[29] Blackbourn, R.L.; Johnson, C.S.; Hupp, J.T. J. Amer. Chem. Soc. 1991, 113, 1060 [ Links ]
[30] Grela, M.A.; Loeb, B.; Restrepo, G.M.; Lagorio, M.G.; San Román, E. In Eliminación de Contaminantes por Fotocatálisis Heterogénea, Libro Colectivo CYTED, M.A. Blesa, Ed., Editorial Digital Graphic, La Plata, 2001 [ Links ]
[31] Gabarell, M.; Chin, Y.P.; Hug, S.; Sulzberger, B. Environ. Sci. Technol., 2003, 37, 4403 [ Links ]
[32] dos Santos Afonso, M.; Morando, P.J.; Blesa, M.A.; Banwart, S.; Stumm W. J. Colloid Interface Sci. 1990, 138, 74 [ Links ]
[33] Alí, S.P.; Blesa, M.A.; Morando, P.J.; Regazzoni, A.E. Langmuir 1996, 12, 4934 [ Links ]

