




![A new building block for polynuclear complexes: the ion [Ru(bptz)(CN)4]2- ( bptz = 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine)](/img/en/next.gif)





Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Asociación Química Argentina
Print version ISSN 0365-0375
An. Asoc. Quím. Argent. vol.92 no.1-3 Buenos Aires Jan./July 2004
REGULAR PAPERS
The Coordination Chemistry Of Nitric Oxide With Pentacyanoferrates
Olabe, J. A.1
1Departamento de Química Inorgánica, Analítica y Química Física, INQUIMAE, Pabellón 2, Ciudad Universitaria, C1428EHA Buenos Aires, Argentina fax: (011) 4576-3341; e-mail: olabe@qi.fcen.uba.ar
Received February 20, 2004. In final form March 25, 2004
Dedicated to Prof. Pedro J. Aymonino on the occasion of his 75th birthday
Abstract
Recent advances in the coordination chemistry studies of the redox-interconverted ligands NO+, NO and NO-(HNO), particularly related to the pentacyanoferrate(II) and (III) fragments, are summarized with mention of structural, spectroscopic (IR and EPR) and DFT calculations. The chemistry of bound NO+ is presented with an emphasis on the mechanisms of the nucleophilic addition reactions of OH-, amines and thiolates on the nitroprusside ion (NP). The reactions of nitric oxide, NO, are addressed through the mechanistic analysis of the formation and dissociation reactions of the [FeII(CN)5NO]3- complex, and through its redox reaction with the [FeIII(CN)5H2O]2- ion, leading to NP. Other biologically significant issues related to the reactivity of bound NO are analysed, namely the disproportionation reactions, the role of dinitrosyls and the identity of nitroxyl, HNO, as a theoretically characterized reduced species, which seemingly appears as an intermediate in redox reactions of small nitrogenated molecules.
Resumen
Se describen avances recientes en la química de coordinación de los ligandos redox-interconvertibles NO+, NO y NO-(HNO), particularmente relacionados con los fragmentos pentacianoferrato(II) y (III), con mención de información estructural, espectroscópica (IR y EPR) y cálculos DFT. Se presenta la química del NO+ coordinado con énfasis en los mecanismos de las reacciones de adición nucleofílica del OH-, aminas y tiolatos en el ion nitroprusiato (NP). Las reacciones del óxido nítrico, NO, se ilustran a través del análisis mecanístico de las reacciones de formación y disociación del complejo [FeII(CN)5NO]3-, y mediante su reacción redox con el ion [FeIII(CN)5H2O]2-, que genera NP. Otros temas biológicamente relevantes relacionados con la reactividad del NO coordinado comprenden las reacciones de desproporcionación, el rol de los dinitrosilos, y la identidad del nitroxilo, HNO, como especie reducida caracterizada teóricamente, la que aparentemente opera como intermediaria en reacciones redox de moléculas nitrogenadas pequeñas.
Introduction and General Background.
Early concerns with the chemistry of nitric oxide (aka nitrogen monoxide, NO) and other reactive nitrogen oxide species were largely focused on their known toxicity as constituents of air pollution.[1] It is now well established that NO plays fundamental roles in biochemical processes, including blood pressure control, neurotransmission and immune response, as well as in tissue damage and carcinogenesis.[2] NO has a ubiquitous place in the chemistry of small nitrogen-containing molecules present in redox cycles in Nature, in the bacterial processes that produce the reversible interconversion of ammonia to nitrite and nitrate in soils.[3] We detail below some of the relevant species important for this type of chemistry, showing the central place of NO.
| Oxid. St. | 3- | 2- | 1- | 0 | 1+ | 2+ | 3+ | 4+ | 5+ |
| N species | NH3 | N2H4 | NH2OH | N2 | N2O, NO-/HNO | NO | NO2-NO+ | NO2 | NO3- |
These redox reactions are frequently metal-catalyzed, as in the case of copper- or iron-based heme-enzymes acting as oxygenases (e.g., NH3 → NH2OH) or as reductases in denitrification reactions (NO2- → NO; NO → N2O; N2O → N2). The mechanisms of these processes are far from disclosed, and are currently under close scrutiny.[3,4]
There is an extensive research activity into the chemistry, biology and pharmacology of NO,[5] and this has led to renewed interest in its fundamental coordination chemistry with the transition metals.[6] In this article we put an emphasis on selected structural and reactivity aspects in the series of [Fe(CN)5L]n- complexes (L may be a versatile ligand),[7] a research area in which Aymonino and collaborators have made significant contributions. The conclusions may be of generalized value for other metal fragments as well, containing aqua, amines, polypyridines, porphyrins and other coligands typical in classical coordination chemistry or in model biomimetic chemistry.[8]
Nitroprusside, [Fe(CN)5NO]2- (NP), first prepared in the middle of 19th century, has a special place among iron-nitrosyl complexes, mainly after the discovery, in 1929, of its effective hypotensive properties. NP is routinely used in clinical studies as an NO-donor drug, although the mechanistic details on the related chemistry are still obscure.[9] It is well known that NO binds to the heme-based guanylate cyclase enzyme in order to promote smooth muscle relaxation and vasodilation, although the signaling mechanism is still under dispute.[10] The chemistry of NP has been early reviewed,[11] and reflects the properties of the nitrosonium (NO+) bound species, namely the reactivity toward nucleophiles (OH-, amines, thiolates, etc.) and its reducibility, which may proceed either through one-electron or multielectronic processes.[12] On the other hand, NP is extremely inert toward thermal dissociation (reflecting the strong Fe-N bond), but it readily releases NO upon visible-near UV photoexcitation, through a redox intramolecular process that presumably leads initially to an FeIII-NO excited-state species.[11]
Upon ligand interchange with NO from NP, the [FeII,III(CN)5H2O]3,2- ions may be produced, and this enlarges the possibilities of studying interesting chemistry related to the coordination and reactivity of most of the small nitrogen-containing species detailed above.[13]
Binding and Redox States of the Nitrosyl Ligand in Cyano-Complexes.
A general feature of nitrosyl-complexes is the delocalized nature of the metal-nitrosyl fragment, {MNO}. Transition metal NO-compounds span variable geometries, coordination numbers and electronic properties due to the differences in electronic configurations of the metal centers and covalent MNO interactions.[6] In the Enemark-Feltham formalism,[14] the complexes are described as {MNO}n, regardless of the orbitals. This description omits the assignment of oxidation numbers to M or NO, although this is frequently ignored by using limiting situations as representative of the electronic structure. NP is an {FeNO}6 species, and the FeIINO+ description is generally accepted, on the basis of spectroscopic and magnetic evidence.[6,11] For octahedral or tetragonal pyramidal geometries, linear or bent arrangements of the {MNO} moieties are predicted for n ≤ or > than 6, respectively. The one-electron reduction product of NP,[Fe(CN)5NO]3-, an {FeNO}7 species, has been known for a long time but has not been structurally characterized.[11] Figure 1 shows the DFT-computed optimized geometries for the linear NP and the reduced 1e- and 2e- bent species.[15]
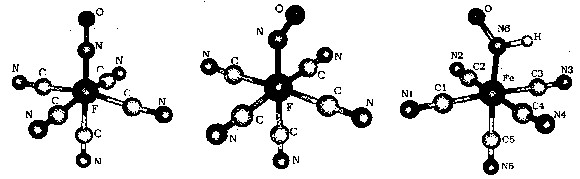
Figure 1. Optimized structures of [Fe(CN)5NO]2-, [Fe(CN)5NO]3- and [Fe(CN)5HNO]3- ions.
These geometrical features are nicely complemented by the IR measurements, given the sensitivity of the IR response to the population of the LUMO, the antibonding {FeNO}orbital (νNO decreases from ca. 1930 to 1650 and 1300-1400 cm-1 upon successive reduction of NP).[15]
Direct characterization of bound HNO in reduced NP has remained elusive, however. Some kinetic evidence exists on its intermediacy during the oxidation of hydroxylamine to NO+,[18] as well as in the complete reduction of NP to ammonia.[19] The chemistry of the HNO ligand is an important current target in modern investigations related to nitrosyl-chemistry.[20]
This section highlights the identification of the linkage isomers of NO achieved during the last decade, namely the side-bound, η2 coordination mode, and the linear, O-bound isonitrosyl (η1 mode), known as the MS2 and MS1 metastable states, respectively.[21]
These species have been first obtained by low-temperature irradiation of NP in the 1970´s, and Aymoninos contributions to their vibrational characterization can be considered crucial, as was also the case for a variety of other nitrosyl-complexes, in the context of a plethora of experimental and theoretical studies.[22,23] The linkage isomers are likely intermediates in chemical and photochemical reactions associated with bound NO, and their potential identification in these situations constitute an open issue for mechanistic studies, which are being extended to the properties of other small molecules, such as N2, N2O, NO2- and so forth (see below).[23]
The Coordination Chemistry of NO with the [FeII,III(CN)5H2O]3,2- Ions.
The formation and dissociation reactions of complexes containing the NO ligand have been very little explored.[4,6,8] Emerging work deals mainly with the coordination ability of NO into iron-porphyrins and related heme-proteins,[24] as well as on other iron-complexes containing polycarboxylate coligands.[8a] In spite of the abundant kinetic and mechanistic work dealing with the substitution chemistry of pentacyano(L)ferrates (II and III, with L = NH3 and amines, py and other N-heterocyclic ligands, etc.),[7] reaction (1) was studied only recently:[25]
Table 1. Selected Bond Distances and Angles (Å and deg), and Infrared Stretching Frequencies (cm-1) for NP and the 1e- and 2e- Reduction Products, [Fe(CN)5NO]3- and [Fe(CN)5HNO]3-, Derived from DFT Calculations (cf. Figure 1)a
|
| [FeII(CN)5(NO+)]2- | [FeII(CN)5(NO)]3- | [FeII(CN)5(HNO)]3- |
| r(FeN6) | 1.62 | 1.74 | 1.78 |
| r(N6-O) | 1.16 | 1.20 | 1.25 |
| r(C-N)av | 1.17 | 1.18 | 1.18 |
| r(Fe-C)av | 1.90 | 1.91 | 1.91 |
| Fe-N-O | 177.2 | 146.6 | 137.5 |
| ν(Fe-N) | 725 | 657 | 808 |
| ν(N-O) | 1932 | 1650 | 1338-1394 |
| ν(C-N) | 2155-2166 | 2000-2084 | 1955-2018 |
a See Reference 15 for the specific Fe-Ci and CiNi values, standard deviations and comparisons with experimental data.
| [FeII(CN)5H2O]3- + NO ¬¾¾® [Fe(CN)5NO]3- + H2O kf, kd | (1) |
A dissociative mechanism has been proposed for the forward formation reaction in (1), as generally found for the pentacyano(L)ferrates(II).[7] The value of kf was 250 M-1 s-1 (T = 25.0 C, I = 0.1 M), very similar to the values obtained for other neutral ligands, in a binding process that is rate-controlled by the dissociation of water. The mechanism is supported by the positive values of the activation parameters, including the volume of activation, with the conclusion that NO behaves as other Lewis-base ligands in the coordination process, without any specific influence of the unpaired electron. A dissociative mechanism also operates for the reverse reaction in (1), with kd = 1.58 x 10-5 s-1(T = 25.0 C, I = 0.1 M, pH 10.2). As shown in Table 2, this is a comparatively low value, suggesting a moderate to strong σ-π interaction of NO with the Fe(II) centre. According to an established correlation between the energies of the d-d absorption bands and the dissociation rate constants,[26] we are now able to place both NO+ and NO ligands with a decreasing strength in the ordering of the spectrochemical series.
Table 2. Dissociation Rate Constants and Activation Parameters for Different [FeII(CN)5L]n- Complexes.a
| Ligand | kd (s-1) | DHd# (kJ mol-1) | DSd# (J K-1 mol-1) | DVd# (cm3 mol-1) |
| NO+ | not detected | - | - | - |
| COb | <10-8 |
|
|
|
| CN- c | ca.4 ´ 10-7 |
|
|
|
| NO | 1.58 ´ 10-5 | 106.4 ± 0.8 | 20 ± 2 | 7.1 ± 0.2 |
| DMSO | 7.5 ´ 10-5 | 110.0 | 46.0 |
|
| Pz | 4.2 ´ 10-4 | 110.5 | 58.6 | 13.0 |
| Py | 1.1 ´ 10-3 | 103.8 | 46.0 |
|
| NH3 | 1.75 ´ 10-2 | 102 | 68 | 16.4 |
a T = 25.0 oC; I = 0.1 M. cf. Reference 25. b Estimated number, measured using pz as a scavenger. c Extrapolated from data reported at higher temperatures.
The low value of kd casts some doubt on the properties of [Fe(CN)5NO]3- as the actual NO-releasing precursor during the activation of guanylate cyclase, given the rapid biological response to NP-stimulation. Other alternatives are being actively investigated (see below).[9]
In contrast to the above picture, NO reacts in a redox-active way in the related reaction (2), which represents a quantitative NO → NO+ conversion, along with iron reduction.[27] Note that the forward path in (2) represents the back, recovery reaction associated with the photochemistry of NP.
| [FeIII(CN)5H2O]2- + NO ¬¾¾® [Fe(CN)5NO]2- + H2O kf, kd | (2) |
The value of kf = 0.25 M-1 s-1 (T = 25.0 C, I = 0.1 M), which is much greater than values usually found for the substitution of other L ligands into [FeIII(CN)5H2O]2- (ca. 10-4-10-7 s-1),[7] together with the activation parameters, support a mechanism as described in Scheme 1. We proposed a rate-limiting reduction of the metal center, followed by nitrite coordination (nitrite is previously generated by fast NO+-hydrolysis) and further conversion to bound NO+. Evidence on the intermediacy of [FeII(CN)5H2O]3- has been provided through competition experiments with pyrazine and NCS- as scavengers. Reaction (2) is an example of the so-called reductive nitrosylation reactions.[4] The small value of kf and the undetectable dissociation in the reverse process can be traced to the low-spin character of the reactant and to the very strong Fe-N bond in NP. A different mechanistic picture was proposed for the ferri-hemes, with a very fast, reversible coordination of NO into the high-spin iron(III), rate-controlled by the water dissociation,[24] and with a product having an electronic structure that is still a matter of discussion, related to the choice of a {FeIINO+}6 or a {FeIIINO}6 description for the heme-nitrosyl product.[4]
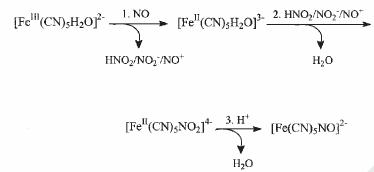
Scheme 1
The Mechanism of the NO+-NO2- Conversion for NP and other NO+-Complexes.
Among the widely studied addition reactions of nucleophiles to NO+-complexes,[12] those comprising OH- as a reactant are in principle very simple mechanistically, because only an acid-base process is involved, as shown in eq (3) for NP:
| [Fe(CN)5NO]2- + 2 OH- ¬¾¾® [Fe(CN)5NO2]4- + H2O | (3) |
It had been proposed early on that reaction (3) proceeds through the attack of a first OH- on the N-atom of NO+ in NP, with formation of bound NO2H as an intermediate-adduct.[28] This adduct may go back to NP or, alternatively, react rapidly with another OH- to form the nitro-complex. A recent DFT calculation on the reaction path in (3) provides the optimized geometries and bonding parameters for the reactant and for the nitrous-acid intermediate and the transition state for the reaction (Figure 2).[29]
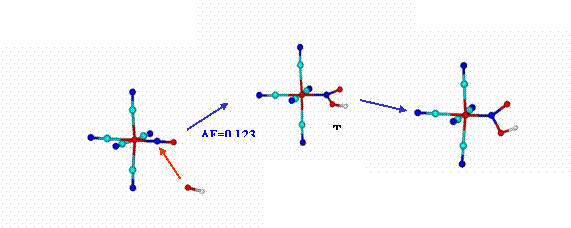
Figure 2. Reaction profile for [Fe(CN)5NO]2- reacting with OH-, rendering the [Fe(CN)5NO2H]3--bound intermediate species. The structures correspond to singular points in the potential hypersurfaces, calculated at a b3lyp-6-31G** level (see more details in Reference 29, including calculation of the TS). Relative energies (y-coordinate) are not drawn to scale. Arrows indicate changes in the molecule that lead to the next step.
In this work, a comprehensive mechanistic study has been pursued for a series of other {X5MNO} complexes. Table 3 displays the measured addition rate constants (representative of the first OH--addition step in eq (3)), and the activation parameters, together with other relevant information, for a selected group of ruthenium-complexes. It can be seen that the rates increase with the redox-potentials of the {RuNO+}6/{RuNO}7 couple, showing greater values for the positively charged complexes (a nice LFER relation is obtained by plotting ln kad against the redox potentials).
Table 3. OH--Addition Rate Constants, Activation Parameters and Corresponding νNO and ENO+NO Values for Different {RuX5NO}n . a
| Compound | kOH (M-s-) b | k3 (s--) c | DH# | DS# | ENO+/NO(V) | nNO(cm-N) |
| cis- | (3.17 + 0.02)×105 | 1.31×105 | 89 + 1 | 159 + 5 | 0.25 | 1946 |
| cis- | (5.06 + 0.02)x104 | 2.75×104 | 83 + 7 | 120 + 20 | 0.18 | 1942 |
| trans- | (1.77 + 0.04)×102 | 9.55×102 | 76 + 2 | 54 + 6 | -0.11 | 1942 |
| trans-[Ru(4-Mepy)(NH3)4NO]3+ | (9.54 + 0.06)×100 | 5.14×100 | 75 + 1 | 26 + 4 | -0.25 | 1934 |
| [Ru(CN)5NO]2- | 9.5 × 10-1 | 6.4 ×100 | 57 | -54 | -0.35 | 1926 |
a Reference 29. b Derived from the rate-law. c Obtained through k3 = kOH/Kip, with Kip being the equilibrium ion-pair formation constant between OH- and the relevant nitrosyl complex, estimated according to an electrostatic model.
Activation enthalpies can be traced to the energy cost of the linear-to-bent conversion in the MNO → MNO2H process, whilst the activation entropies reveal the influence of the charges on the reactants.
Reaction (3) is reversible, and the equilibrium can be perturbed by the ensuing aquation of [Fe(CN)5NO2]4- leading to [Fe(CN)5H2O]3- and free nitrite.[28] We found that the rateof nitrite-aquation reactions differ for the set of complexes in Table 3. The [Fe(CN)5NO2]4- ion releases nitrite readily on a time scale of minutes, but other complexes are much more inert. No systematic kinetic studies are available for these reactions. The formation of bound nitrite starting from reduced substrates such as hydroxylamine and its further delivery from the metal in order to regenerate the active site are key features in the action of the hydroxylamine reductase enzyme.[30] An emerging importance is given to the role of nitrites in biochemistry, particularly highlighted by the recent discovery that deoxyhemoglobin is able to process nitrites reductively leading to NO, with the consequent vasodilatory action.[31]
The Addition Reactions of Other N- and S-Binding Nucleophiles.
These reactions have been also early addressed in the literature.[12] The general mechanistic picture comprises an initial equilibrium between the nitrosyl-complexes and the nucleophiles, with the ensuing decomposition of the adducts, leading to the reduction of NO+ and the oxidation of the nucleophile. The reactions with some N-binding nucleophiles proceed according to the following stoichiometries:[12,19]
| [Fe(CN)5NO]2- + NH2OH + OH- ¾¾® [Fe(CN)5H2O]3- + N2O + H2O | (4) |
| [Fe(CN)5NO]2- + NH3 + OH- ¾¾® [Fe(CN)5H2O]3- + N2 + H2O | (5) |
| [Fe(CN)5NO]2- + N3- + H2O ¾¾® [Fe(CN)5H2O]3- + N2 + N2O | (6) |
| [Fe(CN)5NO]2- + N2H4 + OH- ¾¾® [Fe(CN)5H2O]3- + N2O + NH3 | (7) |
Reaction (7) represents a novel path in which N2O but not N2 is formed as a product of hydrazine oxidation.[19] Scheme 2 shows the mechanism, involving the cleavage of the N-N bond in hydrazine after adduct-formation.

The reaction also provides a catalytic route for the processing of nitrites leading to N2O. The catalyst is the [FeII(CN)5H2O]3- ion, which traps nitrite as NO+ at pH values lower than 10.
Scheme 2 shows the η2- and η1-linkage isomers of N2O, and Figure 3 describes the DFT studies with the calculated geometries for the reactants and intermediates.
The first formation of the η2-isomer is a kinetically controlled process, followed by isomerization to the more stable η1-species and further release of N2O. A comprehensive kinetic and mechanistic study using methylhydrazine, 1,2- and 1,3-dimethylhydrazine allowed the elucidation of different stoichiometries and mechanistic paths according to the structural differences on the nucleophiles.[19]
The detection of linkage isomers has been also achieved for the N2O- and N2-intermediates in reactions (4,5).[32] In addition to the well-known end-on isomer, η1-N2, which has been characterized in other metal centers,[23] the novel η2-N2 coordination mode has been also theoretically predicted, although its high energy precludes its participation in reaction (5) as a true intermediate. The theoretical calculation for reaction (6) predicts that a cyclic adduct-intermediate is accessible prior to the evolution of the mixtures of N2 and N2O.[32]
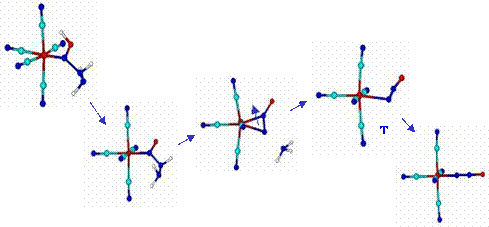
Figure 3. Schematic representation of the calculated stable intermediates formed in initial steps of the reaction of [Fe(CN)5NO]2- with hydrazine, rendering the N2O-bound species. The structures correspond to singular points in the potential hypersurfaces, calculated at a b3lyp-6-31G** level. Relative energies (y-coordinate) are not drawn to scale. Arrows indicate changes in the molecule that lead to the next step. All the adducts and intermediates bear the charge 3-, except the first one containing neutral hydrazine as the added species on [Fe(CN)5NO]2- (this first adduct shows a tautomeric structure, with migration of the vicinal H atom on hydrazine to NO)
As amines are usually protonated at pH values around 7 (with a consequent decrease in nucleophilicity), the mechanistic studies on the addition of thiols and thiolates to bound nitrosyl are considered to be more biologically significant, as also suggested by the role of nitrosothiols as NO-carriers.[4,6,12] The initial adduct-formations evolve as already described for other nucleophiles, as exemplified in reaction (8) for the thiolates, SR-:
| [Fe(CN)5NO]2- + SR- ¬¾¾® [Fe(CN)5(NO)SR]3- | (8) |
In eq (8) the nucleophiles bind through the S-atom to the N-atom of NO+.[33] Kinetic and equilibrium constants have been measured for these reactions at different pH values, showing that the thiolates are stronger nucleophiles than the thiols.[34] The typical intense red colors of the adducts fade for most of the studied thiolates, and this is traced to internal redox decomposition of the nitrosothiolate adduct; this process leads to [Fe(CN)5NO]3- and the thiyl radical, eq (9a), which rapidly dimerizes to the disulfide, eq (9b).[35]
| [Fe(CN)5(NO)SR]3- ¬¾¾® [Fe(CN)5NO]3- + RS· | (9a) |
| RS· + RS· ® RS-SR | (9b) |
Detailed studies have shown that different routes appear when the reactions are studied in excess thiolate, involving complex radical-mediated paths.[36] Thiolate-additions must be studied in anaerobic media, because O2 rapidly converts [Fe(CN)5NO]3- back to NP. In this way, the oxidation of cysteine to cystine by O2 was reported to be catalytically driven when traces of NP are present.[35] The reactions of O2 with NO-bound radicals have not been addressed mechanistically in the literature, in spite of their crucial bioinorganic relevance.[4]
The Disproportionation Reactions of NO-Complexes.
It is well known that gaseous NO slowly disproportionates to N2O and NO2 under ambient conditions, the rate being faster at high pressures, as found in commercial cylinders.[4,6] Catalysis of this process by transition metal complexes has been found in aqueous solutions, with formation of N2O and nitrite, either O- or N-bound to the metal.[4] Active research on these disproportionation reactions is currently in progress employing different complexes, including metal-porphyrins.[4] The available mechanistic proposals comprise either ill defined hyponitrites or dinitrosyl intermediates as precursors of disproportionation.[4] Ongoing work with the [Fe(CN)5NO]3- ion in an excess of NO leads to NP in ca. 50% yield, with other yet unidentified products, suggesting that disproportionation could be operative.[37] Available evidence reveal that dinitrosyl-species could be true intermediates, probably of the {Fe(CN)2(NO)2} type, given the characteristic EPR spectra, also found for other structurally related and biorelevant {FeL2(NO)2} species (L = thiolates, imidazole, etc).[9] The dinitrosyls have been proposed as reactants in transnitrosilation reactions (NO-transfer from a donor to an acceptor, eventually a metalloprotein such as guanylate cyclase).[38] These preliminary results still pose more questions than answers on the detailed mechanism involving the hypotensive role of NP.
Conclusions.
The pentacyanoferrates are useful fragments for the coordination of NO+, as well as of their redox interconverted forms, NO and, potentially, NO- (HNO). The abundant work with NO+-compounds contrasts with the emerging results associated with the NO and HNO ligands, for which reliable structural and spectroscopic characterizations are scarce. Each of these forms shows a specific reactivity toward different reagents (H2O, O2, nucleophiles, light, etc), allowing in many cases for useful comparisons with similar reactions afforded by heme- and non-heme iron enzymes. The nitrosyl-bound species may behave as stable reactants or products, or alternatively as reactive intermediates in the processes associated with the redox interconversions of small nitrogen-containing molecules in natural redox cycles. The [FeII,III(CN)5L]3,2- complexes constitute indeed a valuable benchmark for a systematic study of nitrosyl-chemistry, reinforced by the wide variety of L ligands available for coordination (cf. the N-binding ligands listed above, together with ImH, SH-, SR-, SO32-, CO, etc.). The redox chemistry of most of these ligands is certainly of great bioinorganic relevance.
Acknowledgments.
This work has been supported by CONICET and the University of Buenos Aires, as well as by specific grants from ANPCYT and the Volkswagen Foundation.
References
[1] Swaddle, T. W. Inorganic Chemistry, An Industrial and Environmental Perspective, Academic Press: San Diego, California, 1997. [ Links ]
[2] (a) Lancaster, J., Jr. (Ed) Nitric Oxide, Principles and Actions, Academic Press: San Diego, California, 1996. [ Links ] (b) Feelish, M.; Stamler, J. S., Eds. Methods in Nitric Oxide Research; Wiley: Chichester, 1996. [ Links ] (c) Marnett, L. J., Ed. Chem. Res. Toxicol. 1996, Vol. 9, No. 5. [ Links ]
[3] (a) Averill, B. A. Chem. Rev. 1996, 96, 2951-2964. [ Links ] (b) Wasser, I. M.; de Vries, S.; Moenne-Loccoz, P.; Schroder, I.; Karlin, K. D. Chem. Rev. 2002, 102, 1201, 1234. [ Links ]
[4] Ford, P. C.; Lorkovic, I. M. Chem. Rev. 2002, 102, 993-1018. [ Links ]
[5] (a) Mc Cleverty, J. A. Chem. Rev. 2004, 104, 403-418. [ Links ] (b) Ignarro, L. J.A. Rev. Pharmacol. Toxicol. 1990, 30, 535. [ Links ] (c) Moncada, S.; Palmer, R. M. J.; Higgs, E. A. Pharmacological Reviews, 1991, 43, 109-142. [ Links ]
[6] (a) Richter-Addo, G. B.; Legzdins, P. Metal Nitrosyls; Oxford University Press: New York, 1992. [ Links ] (b) Olabe, J. A.; Slep, L. D. in Comprehensive Coordination Chemistry II, From Biology to Nanotechnology, Mc Cleverty, J. A. and Meyer, T. J., Eds. Vol. 1, Section III, 2003, 603-623, Chap. 1.31. [ Links ] (c) Westcott, B. L.; Enemark, J. H., in Inorganic Electronic Structure and Spectroscopy, Volume II: Applications and Case Studies, Solomon, E. I. and Lever, A. B. P., Eds. 1999, 403-450, Wiley. [ Links ] (d) Stamler, J. S.; Singel, D. J.; Loscalzo, J. Science 1992, 258, 1898. [ Links ]
[7] (a) Sharpe, A. G. The Chemistry of Cyano Complexes of the Transition Metals; Academic Press: London, 1976. [ Links ] (b) Macartney, D. H. Rev. Inorg. Chem. 1988, 9, 101-151. [ Links ] (c) Baraldo, L. M.; Forlano, P.; Parise, A. R.; Slep, L. D.; Olabe, J. A. Coord. Chem. Rev. 2001, 219-221, 881-921. [ Links ]
[8] (a) Wolak, M.; van Eldik, R. Coord. Chem. Rev. 2002, 230, 263-282. [ Links ] (b) Tfouni, E.; Krieger, M.; McGarvey, B. R.; Franco, D. F. Coord. Chem. Rev. 2003, 236, 57-69. [ Links ]
[9] Butler, A. R.; Megson, I. L. Chem. Rev. 2002, 102, 1155-1166. [ Links ]
[10] (a) Ballou, D. P.; Zhao, Y.; Brandisch, P. E.; Marletta, M. A. Proc. Natl. Acad. Sci. USA 2002, 99, 12097-12101. [ Links ] (b) Martí, M.; Scherlis, D. A.; Doctorovich, F.; Ordejón, P.; Estrin, D. A. J. Biol. Inorg. Chem. 2003, 8, 595-600. [ Links ]
[11] (a) Swinehart, J. H. Coord. Chem. Rev. 1967, 2, 385-402. [ Links ] (b) Clarke, M. J.; Gaul, J. B. Struct. Bonding (Berlin) 1993, 81, 147. [ Links ]
[12] Bottomley, F. in Reactions of Coordinated Ligands, Vol. 2, Braterman, P. S., Ed. Plenum Publ. Corp.: New York, 1989. [ Links ]
[13] Olabe, J. A. Adv. Inorg. Chem. 2004, in press. [ Links ]
[14] Enemark, J. H.; Feltham, R. D. Coord. Chem. Rev. 1974, 13, 339-406. [ Links ]
[15] González Lebrero, M. C.; Scherlis, D. A.; Estiú, G. L.; Olabe, J. A.; Estrin, D. A. Inorg. Chem. 2001, 40, 4127-4133. [ Links ]
[16] Wanner, M.; Scheiring, T.; Kaim, W.; Slep, L. D.; Baraldo, L. M.; Olabe, J. A.; Zalis, S.; Baerends, E. J. Inorg. Chem. 2001, 40, 5704-5707. [ Links ]
[17] Masek, J.; Maslova, E. Collection Czechoslov. Chem. Commun. 1974, 39, 2141-2160. [ Links ]
[18] Alluisetti, G. E., Almaraz, A. E.; Amorebieta, V. T.; Doctorovich, F.; Olabe, J. A., to be submitted. [ Links ]
[19] Gutiérrez, M. M.; Amorebieta, V. T.; Estiú, G. L.; Olabe, J. A. J. Am. Chem. Soc. 2002, 124, 10307-10319. [ Links ]
[20] Bari, S. E.; Martí, M. A.; Amorebieta, V. T.; Estrín, D. A.; Doctorovich, F. J. Am. Chem. Soc. 2003, 125, 15272-15273. [ Links ] (b) Sulc, F.; Immoos, C. E.; Pervitsky, D.; Farmer, P. J. J. Am. Chem. Soc. 2004, 126, 1096-1101. [ Links ]
[21] Carducci, M. D.; Pressprich, M. R.; Coppens, P. J. Am. Chem. Soc. 1997, 119, 2669-2678. [ Links ]
[22] Chacón Villalba, M. E.; Guida, J. A.; Varetti, E. L.; Aymonino, P. J. Inorg. Chem. 2003, 42, 2622-2627, and references therein. [ Links ]
[23] Coppens, P.; Novozhilova, I.; Kovalevsky, A. Chem. Rev. 2002, 102, 861-884. [ Links ]
[24] Hoshino, M.; Laverman, L.; Ford, P. C. Coord. Chem. Rev. 1999, 187, 75-102. [ Links ]
[25] Roncaroli, F.; Olabe, J. A.; van Eldik, R. Inorg. Chem. 2003, 42, 4179-4189. [ Links ]
[26] Toma, H. E.; Giesbrecht, E.; Malin, J. M.; Fluck, E. Inorg. Chim. Acta 1975, 14, 11. [ Links ]
[27] Roncaroli, F.; Olabe, J. A.; van Eldik, R. Inorg. Chem. 2002, 41, 5417-5425. [ Links ]
[28] (a) Swinehart, J. H.; Rock, P. A. Inorg. Chem. 1966, 5, 573-576. [ Links ] (b) Masek, J.; Wendt, H. Inorg. Chim. Acta 1969, 3, 455-458. [ Links ]
[29] Roncaroli, F.; Ruggiero, M. E.; Franco, D. W.; Estiú, G. L.; Olabe, J. A. Inorg. Chem. 2002, 41, 5760-5769 [ Links ]
[30] Hendrich, M. P.; Logan, M.; Andersson, K. K.; Arciero, D. M.; Lipscomb, J. D.; Hooper, A. B. J. Am. Chem. Soc. 1994, 116, 11961-11968. [ Links ]
[31] Wink, D. A. Nature Medicine, 2003, 9, 1460-1461. [ Links ]
[32] Olabe, J. A.; Estiú, G. L. Inorg. Chem. 2003, 42, 4873-4880. [ Links ]
[33] Schwane, J. D.; Ashby, M. T. J. Am. Chem. Soc. 2002, 124, 6822-6823. [ Links ]
[34] Johnson, M. D.; Wilkins, R. G. Inorg. Chem. 1984, 23, 231-235. [ Links ]
[35] Morando, P. J.; Borghi, E. B.; Schteingart, L. M.; Blesa, M. A. J. Chem. Soc., Dalton Trans. 1981, 435-440. [ Links ]
[36] Szacilowski, K.; Wanat, A.; Barbieri, A,; Wasiliewska, E.; Witko, M.; Stochel, G.; Stasicka, Z. New J. Chem. 2002, 26, 1495-1502 [ Links ]
[37] Roncaroli, F.; Olabe, J. A., work in progress. [ Links ]
[38] Ueno, T.; Suzuki, Y.; Fujii, S.; Vanin, A. F.; Yoshimura, T. Biochem. Pharmacol. 2002, 63, 485-493. [ Links ]

