










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Asociación Química Argentina
versión impresa ISSN 0365-0375
An. Asoc. Quím. Argent. v.93 n.4-6 Buenos Aires ene./dic. 2005
REGULAR PAPERS
Luminiscence quenching of europium (III) and terbium (III) carboxylates by transition metals in solution
Barja, B. C.; Remorino, A.; Roberti, M. J.**; Aramendia, P.F.*
*INQUIMAE, Departamento de Química Inorgánica, Analítica y Química Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón 2, Ciudad Universitaria, C1428EHA, Buenos Aires, Argentina. Fax: (+54)-11-4576-3341. E-mail: pedro@qi.fcen.uba.ar
**Present address: Max-Planck-Institut für Biophysikalischechemie. Am Fassberg. Göttingen. Germany.
Received May 19th , 2005. In final form August 8th, 2005
Dedicated to the memory of the late Prof. Hans J. Schumacher on the occasion of his 100th birthday
Abstract
The quenching of Eu(NO3)3.6H2O (1), {[Eu3(crot)9(H2O)4].H2O.EtOH}n (2), [Eu2(oda)3.(H2O)2].5H2O}n (3), Na3[Eu(oda)3].2NaClO4.6H2O (4), Na3[Eu(DPA)3].15H2O (5), Tb2(CH3COO)6.2H2O (6), and [Tb(Hoda)3].H2oda (7), where Hcrot (trans CH3-CH=CHCOOH), H2oda (HOOC-CH2-O-CH2-COOH) and H2DPA are crotonic, oxydiacetic, and 2,6- pyridinedicarboxylic acids, respectively, by Cu(II), Ni(II), and Co(II) was studied in aqueous solutions at room temperature. The hydration number of each lanthanide complex was estimated from their excited state lifetime in H2O and in D2O. This value was used to infer the identity of the main species in solution and consequently its electric charge. The dynamic quenching constant shows the influence of the electrostatic interaction. The quenching process is exclusively dynamic for compounds 1 - 4, 6, and 7, while compound 5 shows static and dynamic quenching for the three transition metal ions. The values of the quenching rate constants exceed the energy transfer expectations and show evidence of a collisional mechanism. Specially for compound 4 detection limits for Cu(II) in the range required for drinking water levels are reached.
Resumen
Se estudió la desactivación por Cu(II), Ni(II) y Co(II) en solución acuosa a temperatura ambiente de Eu(NO3)3.6H2O (1), {[Eu3(crot)9(H2O)4].H2O.EtOH}n (2), [Eu2(oda)3.(H2O)2].5H2O}n (3), Na3[Eu(oda)3].2NaClO4.6H2O (4), Na3[Eu(DPA)3].15H2O (5), Tb2(CH3COO)6.2H2O (6) y [Tb(Hoda)3].H2oda (7), donde Hcrot (trans CH3-CH=CHCOOH), H2oda (HOOC-CH2-O-CH2-COOH) y H2DPA son ácido crotónico, oxidiacético, y 2,6- piridindicarboxílico, respectivamente. El número de hidratación de cada complejo de lantánido fue estimado a partir de los tiempos de vida del estado excitado en H2O y en D2O. Este valor fue utilizado para inferir la identidad de las especies mayoritarias en solución y consecuentemente su carga eléctrica. La constante de desactivación dinámica muestra la influencia de la interacción electrostática. El proceso de desactivación es exclusivamente dinámico para los compuestos 1 - 4, 6 y 7, mientras que el compuesto 5 muestra desactivación estática y dinámica para los tres iones de los metales de transición. Los valores de las constantes de velocidad de desactivación exceden los valores esperados para un proceso de transferencia de energía y muestran evidencia de un mecanismo de desactivación colisional. Para el compuesto 4 se alcanzan los límites de detección para Cu(II) requeridos para agua potable.
Introduction
Lanthanide ions display a well-defined luminescence characterized by narrow and highly structured emission bands [1], large difference between absorption and emission wavelengths and lifetimes on the millisecond timescale [2].
The emission wavelength distribution and quantum yield is sensitive to the coordination environment and to the nature of the ligands. In fact, while coordinated ligands can sensitize the lanthanide's luminescence [3,4], the presence of O-H (specially water molecules) or N-H oscillators directly bound to the metal ion provide an efficient pathway for radiationless decay via energy transfer from electronic to vibrational levels [5]. Experiments performed in D2O demonstrate that this quenching effect can lower the luminescence lifetimes by an order of magnitude.
Lanthanide complexes are valuable alternative probes to conventional dyes because the emission is observed in the green-red region of the spectrum where few compounds in natural waters and biological systems emit light. The large difference between excitation and emission wavelengths and their narrow band emission provide the basis for background correction in systems that disperse light.
Lanthanide's long lifetime is not quenched by O2 which makes the detection possible in non degassed media. Additionally their lifetime scale allows time domain separation from the fluorescence emission of naturally emitting species with simple techniques as phosphorescence detection without the drawbacks of phosphorescence (O2 quenching and low yield).
Undoubtedly, lanthanide's luminescent properties make these compounds especially profitable for the design of luminescent labels [6,7,8,9,10] and sensors. In fact, luminescence of Eu(III) and Tb(III) complexes was reported as pH-dependent [11] or as selective anion binding systems in aqueous solution [12]. Transition metal ions are known to be efficient quenchers of the fluorescence emission of organic compounds via dynamic or static mechanisms. Lanthanide complexes can also act as electronic energy transfer sensitizers [13]. This mechanism is also postulated to operate in the emission quenching by transition metals [8,10,14,15]. Nevertheless its contribution to the total quenching events was hard to separate from collisional quenching due to the long emission lifetime of these complexes and to the low absorption coefficient of the transition metal ions in the visible. It is also known that lanthanide chelates are quenched more efficiently by Cu(II) ions than by other fourth period transition metal cations [14,15,16,17].
In this work, quenching measurements were performed for several Eu(III) and Tb(III) complexes by Cu(II), Ni(II), and Co(II) in aqueous solution at room temperature to investigate the influence of the nature of the ligand on the quenching process. Depending on the ligand of each lanthanide complex and on the transition metal quencher involved, only dynamic or both, static and dynamic quenching mechanisms were observed and the quenching constants kQ were evaluated for each case. Luminescence quenching experiments proved to be suitable to detect Cu(II), Ni(II), and Co(II) at concentrations levels lower to the ppm in aqueous solution, a range in which these metal ions are present as pollutants in the environment [18].
Experimental
Transition metal compounds: Copper (II) chloride-dihydrate was obtained from Mallinckrodt, nickel(II) nitrate hexahydrate, from Merck, and cobalt (II) chloride hexahydrate, from Baker Co. They were analytical grade and were used without further purification
Lanthanide compounds. Eu(NO3)3.6H2O 99% (1), was obtained from Fluka Chemie AG, and was used as received. {[Eu3(crot)9(H2O)4].H2O.EtOH}n (2), {[Eu2(oda)3.(H2O)2].5H2O}n(3),Na3[Eu(oda)3].2NaClO4.6H2O(4), a3[Eu(DPA)3].15H2O (5), Tb2(CH3COO)6.2H2O (6) and [Tb(Hoda)3].H2oda.H2O (7) were synthesized according to literature methods [19,20,21,22,23], where Hcrot (trans CH3-CH=CH-COOH), H2oda (HOOCCH2-O-CH2-COOH), and H2DPA are crotonic, oxydiacetic, and 2,6-pyridinedicarboxylic acids, respectively.
Solvents: Water was from a Milli-Q system, D2O (99% D isotopic content) was from Aldrich.
Steady state emission and excitation spectra and quenching measurements
The steady state emission and excitation spectra of all the compounds were recorded on a PTI QuantaMaster QM-1 luminescence spectrometer. Samples were placed in a 1 cm square quartz fluorescence cuvette and measured at right angle geometry. The excitation wavelengths was 394 nm or 532 nm for Eu(III) complexes and 369 nm for Tb(III) complexes for emission spectra. For quenching experiments, the emission wavelengths were 615 nm and 544 nm for Eu(III) and Tb(III) complexes, respectively. Excitation and emission bandwidths were set to 8 and 4 nm, respectively. Excitation spectra were recorded with emission in the 590-595 nm and in the 615 nm bands for Eu(III) and in the 544 nm band for Tb(III) complexes. Excitation and emission bandwidths were set to 4 and 8 nm, respectively. The concentration of the lanthanide complexes was in the 1 - 10 mM range. All experiments were carried out at room temperature at their own pH and ionic force.
Luminescence lifetimes
Samples of Tb(III) and Eu(III) complexes were excited at 354 nm and 532 nm, respectively, with a frequency tripled or doubled Nd:YAG laser (Spectron), which delivered pulses of 8 ns FWHM at 10 Hz and of 30mJ at 354 nm and 150 mJ at 532 nm. These excitation pulses were adequately attenuated before impinging the sample. Samples of compounds 1-7 in H2O or D2O were placed in a 1 cm square quartz fluorescence cuvette and measured at right angle geometry. Emitted light passed through a monochromator and was detected at 488 and 544 nm for Tb(III) samples and at 615 nm for Eu(III) compounds with 4 nm bandwidth. Light was measured with a R928 Hamamatsu photomultiplier and the transient signal was detected by a HP54502 digital oscilloscope and stored on a PC. The traces were fitted to a single exponential decay or to a sum of two exponential terms. The quality of the fit was judged by a homogeneous time-distribution of residuals.
All quenching experiments were performed in air equilibrated solutions. Lanthanide complexes concentrations were in the 1 - 10 mM range.
Results
Steady state emission
Fig. 1 shows the luminescence emission spectra of aqueous solutions of compounds 1 - 5. In all these emission spectra, the characteristic Eu(III) metal centered transition bands (5D0→ 7FJ) are observed, namely for J = 1, 2, and 4 at ca. 590, 615, and 698 nm, respectively The bands for J = 0 and 3 are weak and can only be evident in the figure for some compounds at ca 575 and 650 nm, respectively, though they are present in all complexes. Similarly, Fig. 2 shows the luminescence emission spectra of aqueous solutions of compounds 6 and 7, where the characteristic Tb(III) metal centered transition bands (5D4 → 7FJ) are observed for J = 6, 5, 4 and 3 at ca. 489, 544, 583 and 621 nm, respectively. Excitation spectra showed the typical Eu(III) and Tb(III) bands in the 280-550 nm range, except for 5.

Figure 1. Emission spectra of compounds 1-5 in aqueous solution. Concentrations range from 3 - 5 mM, λexc= 394 nm. Labels show wavelength values of representative spectral features.

Figure 2. Luminescence emission spectra of 5 mM aqueous solutions of compounds 6 and 7, ( λexc= 369 nm).
Degree of hydration of the complexes
Measurements of the luminescent decay times of the compounds 1 - 7 were performed in H2O and D2O to evaluate the mean hydration state of these complexes in aqueous solution. In all cases, the emission decays of the pure complexes were monoexponential in both solvents. Table 1 summarizes the excited state total decay rate constants of the pure compounds in H2O and in D2O as well as the number of coordinated water molecules, q, of each compound. This number was calculated from the first order decay rate constants of the excited state of the lanthanide compounds in H2O (kH2O) and in D2O (kD2O), as described in the literature [5,24], and expressed by Eq. 1. This expression contains corrections to account for the quenching contribution of closely diffusing (second-sphere) water molecules.
| q = A'. ∆kcorr = A' . (kH2O - kD2O + fcorr) | (1) |
where A' = 1.2 ms (5 ms ), and a correction factor fcorr of -0.25 ms-1 (-0.06 ms-1) was applied to∆k for europium (terbium) compounds, respectively.
Table 1. Lifetimes of the luminescence emission in H2O and in D2O, calculated hydration number of the lanthanide ion in solution and dynamic rate constants for the luminescence quenching by Cu(II), by Co(II), and by Ni(II) in H2O of compounds 1 to 7.
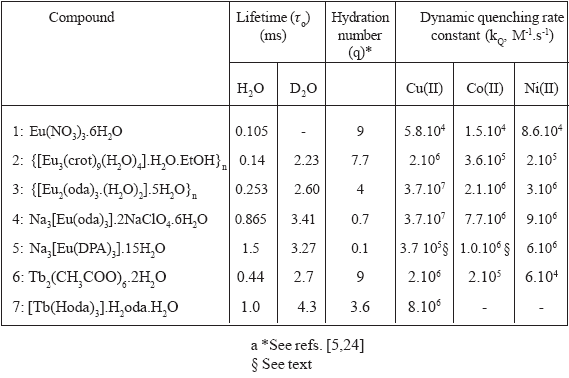
These hydration numbers are used to postulate the identity of the main lanthanide ion containing species in solution, and consequently its charge. The predominant species in solution is dependent on the total concentration of the lanthanide complex added. Our assumptions are restricted to the concentration range in which we performed the measurements: 1-10 mM total complex concentration added.
An hydration number of 9 for compounds 1 and 6 is indicating that the nine coordination sites of the first coordination sphere of the lanthanide ion in solution are all occupied with molecules of water [24]. Consequently the lanthanide containing species in solution is mainly the metal ion with a total charge of +3.
Inversely, a value of q = 0.1 for compound 5 indicates that no coordination site of the first coordination sphere of the Eu(III) is occupied with molecules of water and that three molecules of DPA are bonded to the metal center as is reported in the literature [22]. In order to support this conclusion, we measured the emission spectra of aqueous solutions of Eu(III) and DPA2- at different molar ratios. Figure 3 shows that the addition of DPA2- ligand to Eu(III) alters the emission spectrum, and that the broad band at 590 nm for n = 1 (complex [Eu(DPA)]+) splits into two new ones for n = 3 (complex [Eu(DPA)3]3-). Similar results were observed for the spectra of aqueous solutions of [Tb(DPA)n](3-2.n) with n = 1, 2 and 3 [25]. The carboxylic groups of each ligand help holding the nitrogen in the coordination sphere of the lanthanide ion forming an efficient bridge and thus conferring a high degree of complexation and a total charge of -3 to the predominant Eu(III) species in solution: [Eu(DPA)3]3-.
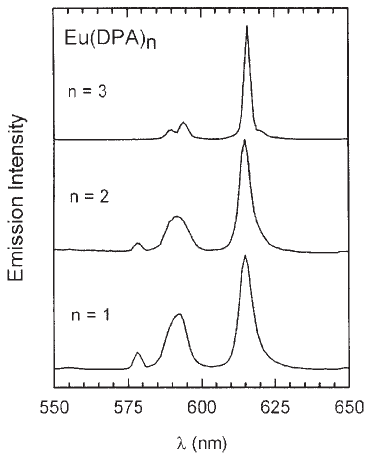
Figure 3. Emission spectra of aqueous solutions of Eu(III) and DPA in different ratios n, where n = moles of ligand / moles of Eu (III). ( λexc= 394 nm).
For the rest of the compounds, the degree of complexation is intermediate, indicating the differences in the strength of the lanthanide ion/ligand bonds. For compound 2, a value of q ≅ 8 indicates that only one site of the coordination sphere of the metal is not occupied by water. Assuming that the carboxylate group is the only functional group capable of coordinating to the Eu(III) center, it is possible to conclude that the predominant Eu(III) species in solution of compound 2 is [Eu(crot)]2+.
When compound 4 is dissolved in water, the Na+ ions are lost and in principle the species [Eu(oda)3]3- could further dissociate into new ones. From crystallographic data, it is known that compound 4 contains the [Eu(oda)3]3- mononuclear species [20] in which the two carboxylate groups and the ether oxygen of each ligand occupy a total of three sites of coordination of the metal centre. According to the value of q = 0.7, it seems reasonable to think that no further dissociation occurs and that the mononuclear nine coordinate [Eu(oda)3]3- remains unaltered in aqueous solution. Similar results were reported for La(III), being the [La(oda)3]3- complex anion the main species present in aqueous solution [26]. It is interesting to note that compounds 3 and 7 have similar values of q even though their crystalline structures are different. Compound 3 is stoichiometrically deficient in ligand to build [Eu(oda)3]3- in solution. Taking into account the high affinity of the lanthanide ion for this ligand, reflected in the q values of 4, it is not unreasonable to assume that [Eu(oda)]+ and [Eu(oda)2]- are present in similar concentrations. Considering that the former can have 6 coordination sites occupied by water, while the later can have 3, the mean value of q would be 4.5, very similar to the value of 4 calculated from the lifetimes. Considering the neat monoexponential decay of the solutions of 3 in both media, we can conclude that the ligand exchange takes place in the submillisecond domain between [Eu(oda)]+ and [Eu(oda)2]-. Compound 7 would have a q = 3 if it either looses a (Hoda)- ligand or if the three (Hoda)- become bidentate ligands in solution. In the first case, the predominant Tb(III) species in solution would have one positive charge, whereas in the second it would be uncharged.
The values of the charge of the predominant lanthanide containing species in solution will be used further to derive conclusions on the electrostatic influence on the dynamic quenching rate constant (see below). Though some of the charge values need further information to be confirmed, they yield a quite consistent picture of the quenching scenario.
Quenching measurements
Luminescence quenching experiments of compounds 1 - 6 by Cu(II), Co(II), and Ni(II) and of compound 7 by Cu(II) were performed in aqueous solution at room temperature. Samples were excited at 394 nm for compounds 1, 2, 3, and 4, at 394 and 532 nm for 5, and at 368 nm for Tb(III) compounds.
Linear Stern-Volmer plots for the steady state data were obtained in all cases (as Figs 4 and 5 show), except for compound 5 where plots with an upper curvature were obtained (see Figs 6 and 7). The dynamic quenching rate constants, kQ, derived from these plots, are summarized in Table 1.

Figure 4. Stern-Volmer plots for the quenching of the emission intensity of compound 3 by Cu (II), Ni (II) and Co (II) in aqueous solution ( λexc= 394 nm and λem = 615 nm). The initial concentrations of the Eu(III) compounds are 5.2, 5.3 and 5.5 mM, respectively.

Figure 5. Stern-Volmer plots for the quenching of the emission intensity of compounds 1- 4 and 6 by Ni (II). The excitation and emission wavelengths are 394 nm and 615 nm for 1- 4 and 369 nm and 544 nm for 6, respectively. The initial concentration of the lanthanide compounds are 5.0, 4.9, 5.5, 5.4 and 2.6 mM in the same order.
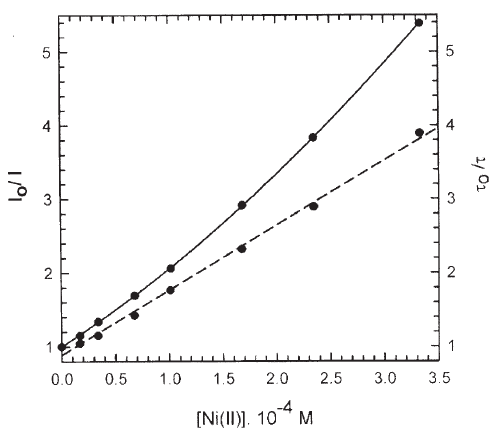
Figure 6. Stern-Volmer plots for the quenching of the emission intensity (solid line) and lifetime (dashed line) of a 4.4 mM aqueous solution of compound 5 by Ni (II). The excitation and emission wavelengths are 532 nm and 615 nm.
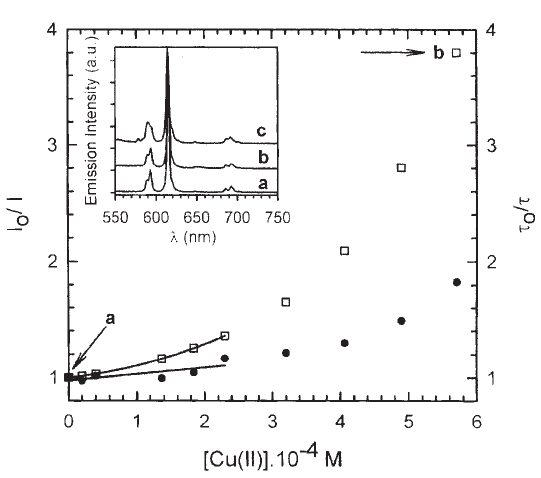
Figure 7. Stern-Volmer plots for the quenching by Cu(II) of the emission intensity (squares) and the longer lifetime of the biexponential emission decay (circles) (See Table 2) of a 2.35 mM aqueous solution of compound 5. The excitation and emission wavelengths are 532 nm and 615 nm for the lifetime data and 394 nm and 615 nm for steady state conditions. The lines correspond to a linear fit to the lifetime data and to a second order polynomial to the total intensity data for [Cu(II)] ≤ 2.30.10-4 M (See Table 2). The inset shows the normalized steady state emission of solutions a and b, marked in the plot, and for a solution c with 1.00 mM total [Cu(II)] added.
For compound 5, the simultaneous presence of static and dynamic quenching for the three cations can be deduced from the fact that the steady state Stern-Volmer plots, Ifo / If , which measure the total quenching, static and dynamic, always show higher quenching ratios than the plots derived from time resolved data, τo/τ (dynamic) for the same quencher concentration. Figs. 6 and 7 show the quenching plots of 5 by Ni(II) and Cu(II), respectively. These plots, performed in the same solution and at the same excitation and emission wavelengths (λexc= 532 nm, λem= 615 nm) were used to discriminate dynamic from static quenching contributions.
For the quenching of 5 by Ni(II), the luminescence decay curves were all monoexponential and a linear Stern-Volmer plot with a slope (equal to kQ.τo) of 8.8 103 M-1 was obtained for the lifetime measurements (Fig. 6). Under steady state conditions, the fit of the curve to a second order polynomial in Ni (II) gave as result an association equilibrium constant, Ka, equal to 1.4 103 M-1 indicating the formation of a 1:1 complex between Ni(II) and compound 5 (Fig. 6) [27]. The fit also renders a value of 8.1 103 M-1 for kQ.τo, in good agreement with the time resolved experiments.
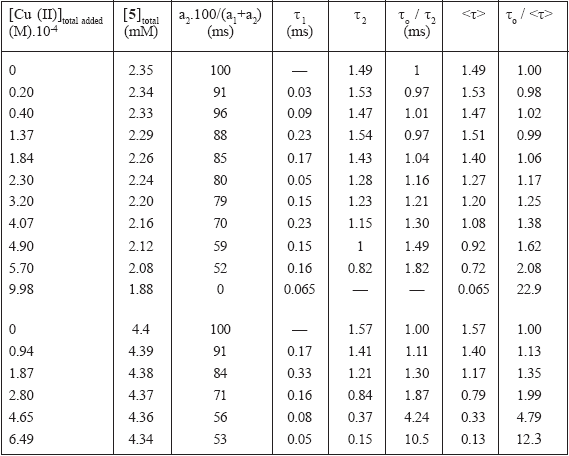
In the case of Cu(II) and Co(II) the behavior is not so simple. The luminescence decay curves showed a clear trend to a biexponential deactivation mode, indicating that perhaps more than one excited state can be involved in the quenching mechanism. Co(II) behaves very similar to Cu(II). Table 2 displays the lifetimes and relative amplitudes of the fit of the luminescence decay of solutions of 5 to a biexponential decay: If = a1.exp(-t/τ1) + a2.exp(-t/τ2), where the τ1,2 are the lifetimes and the a1,2 are the correspondent amplitudes. For both quenchers, the relative amplitude of the faster decay component, a1, increases with transition metal concentration. This results in a decrease in the value of the mean excited state lifetime, <τ> = (a1τ12 + a2τ22) / (a1τ1 + a2τ2). While in the Cu(II) containing system τ1 does not decrease with the transition metal concentration, in the Co(II) system both lifetimes decrease upon Co(II) addition. The luminescence decay in the two solutions with the smaller Cu(II) concentration added can be equally good fitted to a single exponential, thus their shorter lifetime has an uncertainty exceeding 100%. In both cases, τo/<τ>values are always smaller than If0/If values, consistent with the occurrence of static quenching.
Table 2. Lifetimes and relative amplitudes for the biexponential fit of the luminescence decay of compound 5 quenched by Cu(II) and by Co(II) to the equation If(t) = a1.exp(-t/ τ1) + a2.exp(-t/ τ2). < τ> = (a1 τ12 + a2 τ22) / (a1 τ1 + a2 τ2). το = τ2 for the experiments with no added transition metal.
Discussion
The luminescence of all the complexes are efficiently quenched by the three transition metal cations in the sub mM concentration range. The mechanism proposed in the literature for the quenching of lanthanide complexes by transition metals is mainly by electronic energy transfer of the donor lanthanide [8,10,14,15,17,24,28]. The low lying levels of the spin forbidden d-d transitions are directly involved acting as acceptor levels in the lanthanide to transition metal energy transfer mechanism, as reported by Brayshaw et. al. for Co (III) and Cr(III) in the tris- (dipicolinate) rare earth anions [17]. However, this can not fully support the greatest efficiency of Cu(II) as quencher as reported for other Eu(III) complexes [16]. Due to the long lifetime of the lanthanide complexes, the energy transfer process is in the fast diffusion limit [29]. In this case, the quenching process is homogeneous because diffusion averages all donor environments and the second order rate constant for energy transfer quenching takes the form:
|
| (2) |
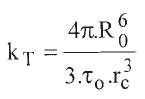
In Eq. 2 R0 is Förster's energy transfer distance (the distance at which the energy transfer frequency to an acceptor equals 1/τo) and rc is the collision encounter distance. Values of R0 for Eu(III) complexes to Co(III) acceptors were estimated by Horrocks and coworkers [10]. Most of them have values of 1.25 ± 0.15 nm. To estimate rc, the hydrated radius of the transition metal ions were calculated from [30] yielding 0.40 nm for Cu(II) and Ni(II), and 0.42 nm for Co(II), while those of the lanthanide complexes were taken from their crystal structures [19,20,21,22,23]. From these values, a sum of radius of 0.9 - 1.2 nm are obtained, the greatest values corresponding to the DPA complexes. If we take an average value of 1.0 nm for rc, we calculate for kT a value of 104 M-1s-1 for lifetimes, τo, of the order of 1 ms. This typical value of kT is much smaller than the values for kQ of Table 1. Values for kQ for Eu(III) complexes by transition metals were reported to be in the 106 - 107 M-1s-1 range [10,16,31,32], in the same order of magnitude as our results. In conclusion, the dipolar energy transfer mechanism makes a small contribution to the overall quenching constants. Thus, in addition to a minor Förster type energy transfer there is a major contribution of a collisional deactivation process (which may include Dexter type energy transfer) [32].
The highest values of kQ are obtained for the more negatively charged species of compounds 3, 4, and 5, while the lowest ones, for the aquo Eu(III) and Tb(III) ions (see Table 1). A direct correlation between kQ and the total charge of the lanthanide species in solution, as derived from the q values, is observed, suggesting that the electrostatic interaction between the lanthanide species and the quencher plays an important role in the dynamic quenching efficiency. The influence of charge interactions in kQ can be described by a linear relationship between ln(kQ) and z1z2e2/ rcεkT, where z1 and z2 are the charges of the lanthanide complex and the transition metal ion, respectively, rc is the collision distance (assumed to be the same for all pairs), ε is the dielectric constant of water and the rest of the symbols have the usual meaning. The plot for the Ni(II) (z2=2) quenching of compounds 1 (z1=3), 2 (z1=2), 3 (z1= 0), 4 (z1=-3), 5 (z1= -3), and 6 (z1=3) renders a good linear relation from which an encounter radius of 1.8 nm is calculated (Figure 8). For Cu(II) and Co(II), the values of rc are in the range 1.7 - 2.3 nm. These values are higher than those calculated from hydrated ionic radii and crystallographic data, as quoted before. The order of magnitude agreement is, nevertheless, considered satisfactory in view of the crude model adopted (for example, no correction for the different ionic strength of the solutions is applied), and evidences the influence of the electrostatic energy in the dynamic quenching rate constant [32].
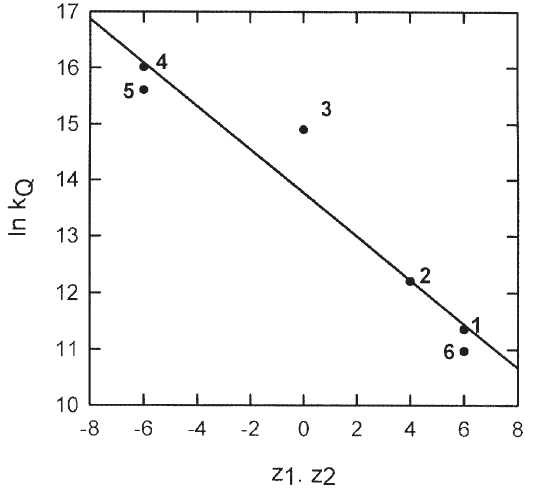
Figure 8. Dependence of the dynamic quenching rate constant kQ on the total charges of compounds 1 - 6 (z1) and the quencher Ni (II) in aqueous solution (z2 = +2) (See text).
From the highest values of the Stern-Volmer constants KSV (M-1) of each cation, it is possible to calculate the minimum concentration values of Cu(II), Co(II), and Ni(II) that yields a 10 % deactivation of the luminescence. From the KSV of compound 4, detection limits of 0.2, 0.7, and 0.9 ppm were calculated for Cu(II), Ni(II), and Co(II), respectively. The value for Cu(II) is satisfactory for traces of this metal to be detected in drinking water, for Ni(II) an order of magnitude improvement would be necessary, while for Co(II) the limit of detection should be improved by two orders of magnitude for this application [18].
The quenching is purely dynamic for compounds 1 - 4, 6, and 7. For compound 5, the overall quenching has similar contributions from static quenching derived from an association of Ni(II) to [Eu(DPA)3]3-, and from a dynamic quenching. The mechanism proposed to explain the results predicts the formation of a ground state complex between the ions, that is surely favored by electrostatic interactions, as well as by the affinity of DPA2- for Ni(II).
The quenching of 5 by Cu(II) and Co(II) has also a static and a dynamic contribution, but the scenario is not so simple as with Ni(II), as evidenced by the biexponential decay of the luminescence and by the nonlinearity of the time resolved Stern-Volmer plots.
It is known from the literature that Cu(II) forms 1:1 and 1:2 complexes with DPA2- as ligand with K1 = 1.4.109 M-1 and K2= 2.4.107 M-1 [33]. Taking into account the corresponding 1:1, 1:2 and 1:3 formation constants for the Eu(III) complexes with DPA2- (K1 =7.108 M-1, K2 = 1.4.107 M-1 and K3 = 3.2.105 M-1) [34], the equilibrium constants for the following reactions are:
| [Eu(DPA)3]3- + Cu2+ ↔[Eu(DPA)2]- + [CuDPA] Keq= 4375 | (3) |
| [Eu(DPA)3]3-+CuDPA↔ [Eu(DPA)2]-+[Cu(DPA)2]2- Keq= 75 | (4) |
So a ligand exchange takes place between Eu(III) and Cu(II). The values of Keq of eqs. 3 and 4 lead to the conclusion that the main species in equilibrium at the concentration ranges of the quenching experiments are [Eu(DPA)3]3- , [Eu(DPA)2]- and [Cu(DPA)2]2-, and that other species in solutions, such as [CuDPA], free Cu(II), free DPA2- and [Eu(DPA)]+, have negligible concentrations.
The inset in Fig. 7 shows the emission spectra of the samples at points a (no Cu(II) added), b, and c for which the molar ratio [Eu(DPA)33-] / [Eu(DPA)2-] are calculated to be 26:1, 0.8:1, and 0.0004:1, respectively. As the concentration of Cu(II) is increased, the fine structure of the band at 590 nm is lost, reflecting that a change in the environment of the emitting species is taking place. We ascribe this, according to Eqs. 3 and 4, to the build up of [Eu(DPA)2]- and [Cu(DPA)2]2- , the last one as the true quencher. Additionally, the spectrum for n = 2 in Fig. 3 closely resembles the spectrum of point c in the inset of Fig. 7. The emission spectrum of point b has comparable contribution from [Eu(DPA)3]3- and [Eu(DPA)2]-. For the quenching behavior this has two consequences. First the [Eu(DPA)2]- complex has a smaller lifetime and emission quantum yield than [Eu(DPA)2]3- because of the replacement of a DPA ligand by three water molecules. Second, the actual quencher is [Cu(DPA)2]2-, with a lower expected kQ than free Cu2+ for both of the Eu(III) species because of its negative charge. The first effect results in a quenching efficiency rapidly increasing with total Cu(II) concentration, which adds to the static and dynamic quenching of each species. Additionally, the absorption coefficients of the two Eu complexes can be different at 532 nm, adding an apparent quenching.
The solution of spectrum c in Figure 7, with a great amount of added Cu(II) and [Eu(DPA)2]- concentration as the predominant emitting species, has a monoexponential decay with 65 µs lifetime.
The simplest way to explain the biexponential behavior is to postulate that the two excited states involved are independently quenched in a dynamic way:
| [Eu(DPA)33-]*+[Cu(DPA)2]2- →[Eu(DPA)3]3-+ [Cu(DPA)2]2- kQ3 | (5) |
| [Eu(DPA)2-]*+[Cu(DPA)2]2- → [Eu(DPA)2]- + [Cu(DPA)2]2- kQ2 | (6) |
As the lifetime of [Eu(DPA)2- ]* is shorter than that of [Eu(DPA)33-]*, the former complex does not deactivate at low Cu(II) concentrations. These conclusions are consistent with the observation of a decreasing longer lifetime with Cu(II) concentration and a constant shorter lifetime. As the complex formation equilibrium rate constants can change in the excited state, a ligand exchange between ground state and excited state Eu(III) complexes can take place. This ligand exchange in the excited state should have increasing importance at higher Cu(II) concentration (that also increases [Eu(DPA)2]- concentration). The reaction represented in Eq. 7 reflects this fact and also acts as a dynamic deactivation of the process
| [Eu(DPA)33-]*+ [Eu(DPA)2]- →[Eu(DPA)3]3- + [Eu(DPA)2-]* | (7) |
At low Cu(II) concentration, the reaction of Eq. 7 cannot efficiently compete with other ways of [Eu(DPA)33-]* deactivation in the ms time range, the decays of the two excited states are independent and it is possible to assign the longer lifetime observed to the decay of [Eu(DPA)33-]* and the shorter to the unquenched decay of [Eu(DPA)2-]*.
In view of the previous discussion, we can make a rough estimation of the static and dynamic quenching parameters for [Eu(DPA)3]3- from the points where the molar ratio [Eu(DPA)33-] / [Eu(DPA)2 -] is high enough and the contribution of the shorter lifetime is less than 20%. Under these conditions, the system can be regarded as containing only [Eu(DPA)3]3- and [Cu(DPA)2]2-. In this way, a value of kQ3.τ0 = 5.6.102 M-1 can be obtained from the linear part of τ2 lifetime Stern-Volmer plot (Fig. 7), from which a value of kQ3 = 3.7 105 M-1.s-1 can be calculated considering the value of τ0 = 1.49 ms (Table 2). From the same plot, an initial slope of 6.2 102 M-1 can be obtained from the total quenching efficiency, which is similar to the value of the dynamic contribution (kQ3.τ0 = 5.6.102 M-1). From the above reasoning, we can conclude that the quenching of [Eu(DPA)3]3- by [Cu(DPA)2]2- is mainly dynamic. The value of kQ3 is similar to the expected one for the deactivation between ions of z1 = -3 and z2 = -2. (See in Table 1 the value of 5.8. 104 M-1.s-1 for the quenching of Eu(III) by Cu(II)). At higher Cu(II) concentration, reaction 7 is important, it contributes to the total deactivation of the complex, and the two lifetimes observed do not directly reflect the decays of each of the two excited states [35].
We can estimate kQ2 for the process of reaction 6 if we take a value of 0.20 ms for the lifetime of the unquenched [Eu(DPA)2]- (an average value of the fast component of Table 2) and we consider the lifetime of 65 µs measured for a solution containing 1.00 mM Cu(II) added (see Table 2). If we assume a linear Stern-Volmer relation, a value of 1.0 107 M-1 s-1 is calculated. This value has the same order of magnitude as the dynamic quenching rate constant of the species [Eu(oda)]+ and [Eu(oda)2]- of compound 3 by Cu(II) (kQ = 3.7.107 M-1.s-1, see Table 1), but is greater than kQ3 because kQ2 corresponds to the quenching of two species with charges of -1 and -2.
In the case of Co(II), the equilibrium constants for the 1:1 and 1:2 complexes with DPA as ligand are K1 = 4.2.106 M-1 and K2 = 1.1.106 M-1 [33].
Similar calculations were performed for the Co(II) / DPA system, being [Eu(DPA)3]3-, [Eu(DPA)2]-, and [Co(DPA)2]2- the main species in equilibrium. Following the same reasoning as in the case of Cu(II), a quenching constant of kQ3 = 1.0. 106 M-1.s-1 was obtained for the dynamic quenching rate constant of [Eu(DPA)3]3- by [Co(DPA)2]2- from the linear portion of the lifetime Stern -Volmer plot. This value is higher than the one expected for the deactivation between ions of z1 = -3 and z2 = -2, however this result can be explained if both the [Co(DPA)2]2- and the neutral [Co(DPA)] complexes are quenching the [Eu(DPA)3]3-species. It can also explain the fact that the shorter lifetime is also quenched by Co(II) (see Table 2).
Finally, for the Ni(II) containing system the equilibrium constants for the 1:1 and 1:2 complexes of Ni2+ with DPA ligand are K1 = 8.9.106 M-1 and K2 = 3.5.106 M-1 [33]. The association constants predict a similar behavior as for the case of Co(II) and Cu(II), which is not observed. We must point out that Ni(II) is a more efficient quencher than Co(II) (see Table 1), so in the Stern Volmer plots, at I0/I = 3.8 [Ni(II)] = 0.23mM (see Figure 6) and the Eu(III):Ni(II) molar ratio is 19:1. This explains why ligand transfer plays no role.
In the case of compounds 3 and 4, the value of the equilibrium constants for the ligand exchange, similar to Eq. 3, with Cu(II), Co(II) and Ni(II) are 10.4, 0.345 and 0.124 [36,37], indicating that ligand exchange is much less favored for oda than it is for DPA. In view of these results, oda seems to be the most suitable ligand, among all those studied in this work, to detect Cu(II), Co(II) and Ni(II) by means of quenching experiments. In particular, compound 4 due to its long excited state lifetime, possesses the highest values of Stern-Volmer constants for the quenching of these transition metals (of the order of 104 M-1), which renders the highest sensitivity to the method. The lack of static quenching together with the linear plots obtained in all the experiments makes compound 4 very simple to work with.
Acknowledgements
PFA is a member of Carrera del Investigador Científico (Research Staff) from CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina). The work was supported by grants PID 0388 (CONICET), PICT 06-04438 (ANPCyT, Argentina), and Fundación Antorchas (Argentina). We thank Dr. Mireille Perec for provision of some compounds and helpful discussions.
References
[1] Ofelt, G. S., J. Chem. Phys., 1963, 38, 2171-2180. [ Links ]
[2] Parker, D.; Gareth Williams, J. A., Chem. Soc. Dalton Trans., 1996, 3613. [ Links ]
[3] Quici, S.; Marzanni, G.; Cavazzini, M.; Anelli, P. L.; Botta, M.; Gianollo, E.; Accorsi G.; Armaroli, N.; Barigelletti, F., Inorg. Chem., 2002, 41, 2777. [ Links ]
[4] Lowe, M. P.; Parker, D., Inorg. Chim. Acta. 2001, 317, 163. [ Links ]
[5] Beeby, A.; Clarkson, I. M.; Dickins, R. S.; Faulkner, S.; Parker, D.; Royle, L.; de Sousa, A. S.; Gareth Williams, J. A.; Woods, M.; J. Chem. Soc. Perkin Trans., 1999, 2, 493. [ Links ]
[6] Richardson, F.S., Chem Rev., 1982, 82, 541-552. [ Links ]
[7] Martin, R. B.; Richardson, F. S., Q. Rev. Biophys., 1979, 12, 184. [ Links ]
[8] Bruno, J.; Horrocks, W. DeW.; Zauhar, R. J., Biochemistry, 1992, 31, 7016. [ Links ]
[9] Tomas, D. D.; Stryer, L., J. Mol. Biol., 1982, 154, 145. [ Links ]
[10] Cronce, D. T.; Horrocks, W. DeW., Biochemistry, 1992, 31, 7963. [ Links ]
[11] Blair, S. M.; Lowe, P.; Mathieu, C. E.; Parker, D.; Kanthi Senanayade, P.; Kataky, R., Inorg Chem, 2001, 40, 5860. [ Links ]
[12] Montalti, M.; Prodi, L.; Zaccheroni, N.; Charbonniere, L.; Douce, L.; Ziessel, R., J. Am. Chem. Soc., 2001, 123,12694. [ Links ]
[13] Selvin, P. R.; Rana, T. M.; Hearst, J. E., J. Am. Chem. Soc., 1994, 116, 6029. [ Links ] Selvin, P. R., Nature Struct. Biol., 2000, 7, 730. [ Links ]
[14] Bünzli, J. C. G.; Piguet, C., Chem. Rev., 2002, 102, 1897-1928. [ Links ]
[15] Horrocks Jr., W. DeW.; Holmquist, B.; Vallee, B. L., Proc. Nat. Acad. Sci. USA, 1975, 72, 4764. [ Links ]
[16] Kessler, M. A., Anal. Chim. Acta, 1998, 364, 125. [ Links ]
[17] Brayshaw, P. A.; Bünzli, J. C. G.; Froidenaux, P.; Harrowfield, J. M.; Kim, Y.; Sobolev, A. N., Inorg. Chem., 1995, 34, 2068. [ Links ]
[18] These values are 1.3, 0.1, and 0.05 ppm for Cu(II), Ni(II) and Co(II) according to the maximun EPA metal contaminant levels for drinking water.
[19] Barja, B. C.; Aramendia, P.F.; Baggio, R.; Garland, M. T.; Peña, O.; Perec, M., Inorg. Chim. Acta, 2003, 355, 183. [ Links ]
[20] Aramendia, P.F.; Baggio, R.; Garland, M. T.; Perec, M., Inorg. Chim. Acta, 2000, 303, 306. [ Links ]
[21] Albin, M.; Whittle, R. R.; Horrocks Jr., W. DeW., Inorg.Chem., 1985, 24, 4591. [ Links ]
[22] Murria, G. M.; Sarrio, R. V.; Peterson, J. R., Inorg. Chim. Acta, 1990, 176, 233. [ Links ]
[23] Barja, B.; Baggio, R.; Garland, M.T.; Aramendia, P.F.; Peña, O.; Perec, M., Inorg. Chim. Acta, 2003, 346C, 187. [ Links ]
[24] Horrocks Jr., W. DeW.; Sudnick, D. R., J. Am. Chem. Soc., 1979, 101, 334. [ Links ]
[25] Dechter, J. J., Progr. Inorg. Chem.,1983, 33, 393. [ Links ]
[26] Lakowicz, J. R., Principles of Fluorescence Spectroscopy, 2nd Edition, Kluwer Academic/ Plenum Publishers, New York, 1999, 239-343. [ Links ]
[27] Horrocks Jr., W. DeW.; Tingey, J. M., Biochemistry, 1988, 27, 413. [ Links ]
[28] Thomas, D. D.; Caslsen, W. F.; Stryer, L., Proc. Natl. Acad Sci U. S. A., 1978, 75, 5746. [ Links ]
[29] Marcus, Y., Ion Solvation ed. John Wiley & Sons, New York, 1985, 96-105. [ Links ]
[30] Metcalf, D. H.; Stewart, J. M. M.; Snyder, S. W.; Grisham, C. M.; Richardson, F. S., Inorg. Chem., 1992, 31, 2445. [ Links ]
[31] Metcalf, D. H.; Bolender, J P.; Driver, M S.; Richardson, F. S., J. Phys. Chem., 1993, 97, 553. [ Links ]
[32] Andereg, G., Helv. Chim. Acta, 1960, 54, 414. [ Links ]
[33] Grenthe, I., J. Am. Chem. Soc., 1963, 83, 360. [ Links ]
[34] Jones II, G.; Vullev, V. I., Photochem. Photobiol. Sci., 2002, 1, 925. [ Links ]
[35] Bernasconi, C. F., Techniques of Chemistry John Wiley & Sons, 1986, 425-485. [ Links ]
[36] Grenthe, I.; Tobiasson, I. Acta Chem. Scand., 1963, 17, 2101. [ Links ]
[37] Arena, G.; Musumeci, S.; Rizzarelli, E.; Sammartano, S. Inorg.Chim. Acta, 1978, 27, 31. [ Links ]

