










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Asociación Química Argentina
Print version ISSN 0365-0375
An. Asoc. Quím. Argent. vol.93 no.4-6 Buenos Aires Jan./Dec. 2005
REGULAR PAPERS
Thermal Gas-Phase Oxidation Of Trifluorobromoethene, CF2CFBr, Initiated By NO2
Arce, V.a; dos Santos Afonso, M.b; Romano, R.M.a; Czarnowski, J.c
aCEQUINOR (CONICET, UNLP) Departamento de Química, Facultad de Ciencias Exactas, Universidad de La Plata, 47 y 115, 1900 La Plata, Argentina.
bINQUIMAE, Departamento de Química Inorgánica, Analítica y Química, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón II, 3er Piso, (1428) Buenos Aires, Argentina.
cInstituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas, INIFTA, Sucursal 4, Casilla de Correo 16, (B1906ZAA) La Plata, Argentina
Fax: +54 221 425 4642, Email: bdq782@infovia.com.ar
Received July 26th, 2005. In final form September 2nd, 2005
Dedicated to the memory of the late Prof. Hans J. Schumacher on the occasion of his 100th birthday
Abstract
The oxidation of CF2CFBr by molecular O2 initiated by the addition of NO2 to the double bond of the alkene was studied at 313.4 K, using a conventional static system. The initial pressure of CF2CFBr was varied between 18.8 and 43.9 torr, that of NO2 between 0.9 and 4.8 torr and that of O2 between 96.9 and 402.9 torr. The following products were formed: CF2BrC(O)F, as the main product, C(O)F2 and C(O)FBr and small amounts of peroxynitrate, CF2BrCFBrO2NO2, and trifluorobromoethene epoxide.
CF2BrC(O)F was characterized by its IR spectrum consistent with both the proposed structure and the calculations carried out using ab initio and Density Functional Theory methods. In presence of CF2CFBr the reaction proceeded with a pressure decrease. After the alkene was consumed an increase of the pressure and formation of bromine was observed. The oxidation is a chain reaction of pseudo-zero order with respect to O2 as reactant at the pressure of oxygen used in this work. Its basic steps are: chain initiation by addition of NO2 to the double bond leading, through reaction sequence in presence of O2, to generation of bromine atoms and chain propagation by reaction of Br• with alkene, originating CF2BrCFBrO2• and CF2BrCFBrO• radicals. The predominant fate of the latter is the bromine atoms extrusion, with C-C bond cleavage playing only a minor role. A full mechanism is postulated. The value of (2.2 ±1) x 10-5 s-1 was obtained for the room temperature rate constant for the unimolecular decomposition of CF2BrCFBrO2NO2.
Resumen
Se estudió la oxidación de CF2CFBr por O2 molecular iniciada por la adición de NO2 al doble enlace del alqueno a 313,4 K en un sistema convencional estático. Se varió la presión inicial de CF2CFBr entre 18,8 y 43,9 torr, la de NO2 entre 0,9 y 4,8 torr y la de O2 entre 96,9 y 402,9 torr. Se formaron los siguientes productos: CF2BrC(O)F, como producto principal, C(O)F2 y C(O)FBr y pequeñas cantidades de peroxinitrato, CF2BrCFBrO2NO2 y epóxido de trifluorobromoeteno. Se caracterizó el CF2BrC(O)F por su espectro infrarrojo comparándolo con el calculado usando métodos ab initio y la teoría de los funcionales de la densidad. En presencia de CF2CFBr la reacción procede con disminución de la presión. Consumido el alqueno se observó aumento de la presión y la formación de bromo. A las presiones de oxígeno usadas en este trabajo, la oxidación es una reacción en cadena de seudo-cero orden con respecto a O2 como reactivo. Sus pasos básicos son: iniciación de cadena por adición de NO2 al doble enlace, que en presencia de O2, mediante una secuencia de reacciones, genera los átomos de bromo y la propagación de la cadena por reacción de Br• con el alqueno, originando radicales CF2BrCFBrO2• y CF2BrCFBrO•. Este último se descompone principalmente por eliminación de átomo de bromo y en menor grado por la ruptura de enlace C-C. Se postuló el mecanismo completo de la reacción. Se obtuvo el valor de (2,2 ±1) x 10-5 s-1 para la constante de descomposición unimolecular de CF2BrCFBrO2NO2 a temperatura ambiente.
Introduction
In the reactions between NO2 and halogenated olefins, NO2 can act as nitrating or oxidating agent. The products of these reactions are nitrohaloacetyl halides and XNO, where X = Cl or F, vicinal dinitro products and nitrohaloketones [1-8]. These works were generally executed for preparative purposes.
The detailed kinetic and mechanistic studies were reported for the thermal gas-phase reactions of NO2 with CF2CF2 [9], CF2CFCl, [10], CF2CCl2 [11], CClHCCl2 [12], perfluoropropene, C3F6, [13] and CF2CFBr [14]. In these works a basic common reaction mechanism for nitration or oxidation of CX2CX2 by NO2, where X = Cl, F or Br, can be postulated as follows:
| 1. | NO2 + CX2CX2 → CX2(NO2)CX2• |
| 2. | CX2(NO2)CX2• + NO2 + M → CX2(NO2)CX2NO2 + M |
| 3. | CX2(NO2)CX2• + NO2 → CX2(NO2)C(O)X + XNO |
The products of nitration of CF2CF2 were CF2(NO2)C(O)F and FNO, those of CF2CFCl were CF2(NO2)CFClNO2, CF2(NO2)C(O)F and ClNO and those of CF2CCl2 were CF2(NO2)CCl2NO2, CF2(NO2)C(O)Cl and ClNO. In the reaction of NO2 with trichloroethene, NO2 oxidizes CClHCCl2, giving HC(O)C(O)Cl and ClNO as the only products [15]. The main products of the reaction between NO2 and C3F6 were the oxidation product perfluoropropene oxide (PFPO) and nitric oxide, NO, produced in equivalent amounts. The dinitro-compound CF3CF(NO2)CF2NO2 and nitroperfluoroacetone CF3C(O)CF2NO2 were also formed in minor amounts. The relation R= perfluoropropene oxide]/ ([CF3CF(NO2)CF2NO2]+[CF3C(O)CF2NO2]) increased with temperature. In the reaction of NO2 with CF2CFBr the observed products were CF2(NO2)C(O)F, CF2(NO2)CFBrNO2, BrNO, Br2 and NO.
The addition of O2 to the reaction system NO2 + 1,1-dichlorodifluoroethene, changed the reaction course [16]. The oxidation of alkene, initiated by the addition of NO2 to the double bond of the olefin, occurred, leading in presence of O2 to the formation of nitroperoxy-radicals, CF2(NO2)CCl2O2• and nitrooxy- radicals, CF2(NO2)CCl2O•. The CF2(NO2)CCl2O• radicals released chlorine atoms, which add to 1,1-dichlorodifluoroethene originating radicals CF2ClCCl2O2• and CF2ClCCl2O•. The free chlorine atoms generated from CF2ClCCl2O• radicals initiate a chain reaction with chlorine atoms as chain carriers, giving haloacetyl chloride, CF2ClC(O)Cl as the main product. Some 15% of the CF2ClCCl2O• radicals decomposed by the scission of their C-C bond leading to the formation of C(O)F2 and C(O)Cl2 and reforming chlorine atoms. Also small amounts of peroxynitrate CF2ClCCl2O2NO2 and 1,1- dichlorodifluoroethene epoxide were formed.
The NO2-initiated oxidation of tetramethylene was reported in literature [17]. The use of chemical initiators in absence of light permits a better control of the reaction course in organic synthesis. The stable and easily handled trifluoromethyl hypofluorite, CF3OF, containing a weak O-F bond (43.5 kcal/mol) [18-20] is also an effective initiator of oxidation of haloalkenes (E). CF3OF adds to the double bond giving CF3O(E) radicals, where E = CF2CCl2 [21], CHClCCl2 [22] and CCl2CCl2 [23]. The reaction of these radicals with O2 is very fast leading to the formation of CF3O(E)O, which principally decomposes by the detachment of the Cl from the CCl2O group of the alkene. The chlorine atoms react rapidly with CX2CCl2, where X = H, Cl or F, initiating in presence of O2, a chain reaction with Cl• as chain carrier and giving CX2ClC(O)Cl as the main product. Minor amounts of C(O)X2, where X = H, Cl and F, are also formed. In the case of C(O)HCl, it decomposes rapidly to CO and HCl.
The direct Cl- and Br-atom initiated oxidation of CHClCCl2 [24,25] and CCl2CCl2 [25] and Br-atom oxidation of CHClCCl2 and CCl2CCl2 [26] were reported. These studies were made to investigate the atmospheric fate of trichloro- and tetrachloroethenes, used as solvents, dry cleaning agents and degreasing agents and released to the atmosphere in 90-100 % of their anthropogenic production.
The basic common chain reaction mechanism for the oxidation of haloalkenes, initiated by the addition of NO2 or CF3OF to the double bond of olefins in presence of O2, and that initiated by ultraviolet or visible photolysis of Cl2 and Br2, can be resumed as:


In this work the investigation of the reaction of NO2 with bromotrifluoroethene, CF2CFBr, in presence of molecular oxygen was undertaken to elucidate the elementary steps involved and to characterize the products. Comparing with the analogue reaction between NO2 and chloroperfluoroalkene, CF2CFCl in presence of oxygen [11], where free chlorine atoms are produced, it is reasonable to postulate that the free bromine atoms are formed in the reaction system NO2 + CF2CFBr + O2. We have found no data on the addition of bromine atoms to fluorinated olefins.
Experimental
All reactants were purchased commercially. NO was eliminated from NO2 (Matheson 99.5 %) by a series of freeze-pump-thaw cycles in presence of O2 until disappearance of the blue colour of N2O3. Finally, the degassed NO2 was purified by fractional condensation using the fraction that distilled between 213 and 243 K. The CF2CFBr (PCR, 97-98%) contained CF4, and CF3CF3 as impurities. These impurities are more volatile than CF2CFBr, but could not be separated by fractional condensation, distilling together. The CF2CFBr was purified by intermittent brief evacuation cycles at 153 K, opening and closing the trap valve. This procedure was repeated several times, until the disappearance of the respective very strong absorption bands of CF4 [27] and CF3CF3 [28] at 1279 and 1250 cm-1 in the IR spectrum of CF2CFBr. Oxygen was bubbled through 98 % analytical-grade H2SO4 and passed slowly through a Pyrex coil at 153 K.
The experiments were performed in a grease-free static system, allowing pressure measurements at constant volume and temperature. A spherical quartz bulb of 270 cm3 (S/V = 0.75 cm-1) was used as reaction vessel. The pressure was measured with a quartz spiral gauge and the temperature maintained within ±0.1 K using a Lauda thermostat.
The reaction was followed measuring the pressure change as a function of time. 21 experiments were made at 313.4 K. The initial pressure of CF2CFBr was varied between 18.8 and 43.9 torr, that of NO2 between 0.9 and 4.8 torr and that O2 between 96.9 and 402.9 torr. Initially the reaction proceeded with pressure decrease. After all CF2CFBr was consumed a slow pressure increase was observed.
Infrared spectra of the reaction mixtures were recorded on a Shimadzu IR-435 spectrometer and a Perkin-Elmer 1600 Series FTIR spectrometer, using a 10 cm cell provided with NaCl and KBr windows, respectively. The gas FTIR spectra of the products, separated by fractional condensation after all alkene was consumed, were recorded on a Nexus Nicolet instrument equipped with a cryogenic MCTB detector between 4000 and 400 cm-1 at room temperature, using a 10 cm cell provided with KBr windows. The UV-visible spectra of the products in the gas phase were recorded on a Hewlett-Packard Model 8452A spectrometer, using a 10 cm quartz cell.
Results
In the presence of CF2CFBr the reaction proceeded with a pressure decrease, -∆p/∆t. After all the alkene was consumed the reaction continued with a slow pressure increase, ∆p/∆t, as a function of time. No reaction between CF2CFBr and O2 was observed after several hours in the absence of NO2.
The following products were formed: CF2BrC(O)F as the main product, minor quantities of C(O)F2 and C(O)FBr, and small amounts of CF2BrCFBrO2NO2 and trifluorobromoethene epoxide (TFBrEO). The compound CF2BrC(O)F was characterized by its IR spectrum consistent with the gauche structure and with the calculations for this molecule using ab initio and Density Functional Theory methods [29]. The identification was confirmed by comparison with the experimental IR spectrum of CF2ClC(O)F [30], given that infrared spectra of analogous molecules, where the bromine atom is substituted by chlorine are similar. The most intense absorptions for this molecule are found to appear at 1887, 1195, 1102 and 937 cm-1, and are assigned to the ν(CO), νas(CF2), νs(CF2), and ν(CF), respectively. The compounds C(O)F2 [31] and C(O)FBr [32,33] were identified by their respective IR spectra and the formation of C(O)FBr was also detected by its UV spectrum at the range of 200-220 nm [33]. The product CF2BrCFBrO2NO2 was identified by its infrared absorption band at 1758 cm-1, characteristic to the NO2 group of the haloalkylperoxynitrates and haloalkoxylperoxynitrate. This band appears at 1754 cm-1 for CCl3O2NO2 [34-36], at 1757 cm-1 for CCl2FO2NO2 [34-36], at 1761 cm-1 for CClF2O2NO2 [34,35], at 1762 cm-1 for CF3O2NO2 [37], at 1761 cm-1 for CF3C(O)O2NO2 [38], at 1759 cm-1 for CClF2C(O)O2NO2 [38], at 1755 cm-1 for CCl2FC(O)O2NO2 [38], at 1755 cm-1 for CCl3C(O)O2NO2 [38] and 1758 cm-1 for CF2ClCCl2O2NO2 [16]. The trifluorobromoethene epoxide (TFBrEO), was identified in the reaction mixture by its infrared band at 1540 cm-1, assigned to the ring-breathing mode, characteristic of fluoroepoxides. This band appears at 1500 cm-1 for 1,1-dichloro-2.2-difluoroethene epoxide and chlorotrifluoroethene epoxide [39] and at 1551 cm-1 for perfluoropropene epoxide [13].
After the complete consumption of CF2CFBr, the reaction continued with pressure increase, ∆p/∆t, producing Br2 and reforming NO2 in addition to CF2BrC(O)F, C(O)F2 and C(O)FBr. The bromine was identified by its UV spectrum [40].
All experiments were carried out to the complete consumption of CF2CFBr. For analyzing the reaction mixtures the reaction vessel was rapidly cooled to liquid air temperature and the mixture separated by fractional condensation. When the reaction was interrupted at the inflection point, when -∆p/∆t changed to ∆p/∆t, the first fraction Fr1, volatile at 158 K, consisted of C(O)F2 and C(O)FBr, the second fraction Fr2, volatile at 183 K, was CF2BrC(O)F and the third fraction Fr3 remaining as a residue at 158 K consisted of NO2 and CF2BrCFBrO2NO2. In the infrared spectrum of this fraction, weak absorption bands at 1321, 1025 and 821 cm-1 were observed. These bands are characteristic of the symmetric stretching of NO2 in the C-NO2 group and of the C-F and C-N groups, respectively, and were attributed to the presence of traces of CF2BrCFBrNO2. The very strong band at 1618 cm-1 corresponding to the asymmetric stretching of NO2 in the C-NO2 group was overlapped by the broad band of NO2 at 1635- 1590 cm-1, suggesting that CF2BrCFBrNO2 was formed in very small amounts. When the reaction was interrupted while it proceeded with the pressure increase, the fractions Fr1 and Fr2 remained unchanged, but the fraction Fr3 contained Br2 in addition to CF2BrCFBrO2NO2, NO2 and CF2BrCFBrNO2.
The absence or presence of Br2 in the reaction mixtures at different reaction time intervals corresponding to the pressure decrease and pressure increase, respectively, was monitored by UV spectroscopy.
It was observed in the successive infrared spectra of the same fraction Fr2, that intensity of the absorption band of CF2BrCFBrO2NO2 at 1758 cm-1 decreases as a function of time, appearing bands of CF2BrC(O)F and very small bands of C(O)F2 and C(O)FBr, and increasing the intensity of the band of NO2. In addition, the formation of Br2 was detected by its UV spectra.
Considering that at low pressure the infrared absorption is proportional to the concentration of the corresponding compound, the value of (2.2±1) x 10-5 s-1 was obtained for k, the effective room temperature rate constant for the unimolecular decomposition of CF2BrCFBrO2NO2, measuring the absorbance A at 1758 cm-1 as a function of time t and using k= ln (Am / An) / (tm - tn).
The reaction is a homogeneous chain reaction whose rate depends on the relation between NO2, CF2CFBr and O2 and the total pressure. At the pressures of O2 used in this work, the reaction is of the pseudo-zero order with respect to O2 as reactant, indicating the third body character of O2. The initial reaction rates were proportional to the respective pressures of NO2 and CF2CFBr and also were influenced in a specific way by the total pressure. At pressure of NO2 and the temperature of our work, the equilibrium constant for N2O4 ↔ 2 NO2 [41] indicated that N2O4 was practically dissociated into NO2.
The behavior of the reaction rate, -∆p/∆t, as a function of time, corresponding to the pressure decrease interval, is illustrated in Figs. 1-3. The initial pressures of NO2, CF2CFBr and O2 in torr for each run presented are given inside the graphs. The analytical data of 10 experiments are summarized in the Table 1, where indices i and f signify initial and final, respectively.
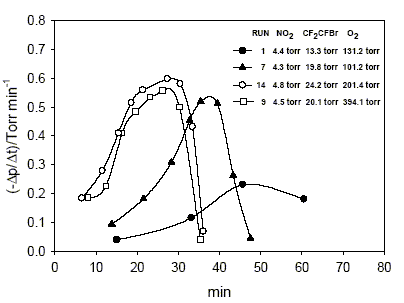
Fig. 1. Reaction rates, - ∆p/ ∆t as a function of time, corresponding to a pressure decrease interval. Initial pressures of NO2, CF2CFBr and O2 in torr for each run are given inside the graph.

Fig. 2. Reaction rates, - ∆p/ ∆t as a function of time corresponding to a pressure decrease interval. Initial pressures of NO2, CF2CFBr and O2 in torr for each run are given inside the graph.

Fig. 3. Reaction rates, - ∆p/ ∆ as a function of time t, corresponding to a pressure decrease interval. Initial pressures of NO2, CF2CFBr and O2 in torr for each run are given inside the graph.
Table 1. Analytical data of 10 experiments carried out to the complete consumption of CF2CFBr.


Discussion
In order to propose the reaction mechanism, in addition to products analysis, the following reactions were considered: the reactions between NO2 and CF2CFCl [10], CF2CCl2 [11], CHClCCl2 [12], and CF2CFBr [14] in absence of O2, the reaction of NO2 with CF2CCl2 in presence of O2 [16], the oxidation of CF2CCl2 [21], CHClCCl2 [22] and CCl2CCl2 [23] initiated by CF3OF addition to the double bond, the bromine and chlorine atoms initiated oxidation of CHClCCl2 [24,25] and CCl2CCl2 [25] and the respective oxidations of CHClCCl2 and CCl2CCl2 [26] initiated by the bromine atoms.
The following mechanism appears to be consistent with the experimental results obtained in our work:


Other steps were also considered: CF2(NO2)CFBr• + NO2 + M → CF2(NO2)CFBrNO2 + M and CF2(NO2)CFBrO2• + NO2 + M → CF2(NO2)CFBrO2NO2 + M but, as their products have not been detected, these reaction can be neglected in comparison with the steps of the chain reaction. Besides, they did not appear necessary to explain the results.
The primary path is the thermal addition of NO2 to CF2CFBr, giving CF2(NO2)CFBr• radicals. The corresponding rate constant for this reaction, k1 = (1.51±0.45) x 106 exp(-(10.88±1) kcal mol-1/RT) dm3 mol-1 s-1, was determined in a previous work [14]. The oxidation of CF2(NO2)CFBr• radicals leads through reactions (2) and (3) to the formation of CF2(NO2)CFBrO• radicals, which decompose giving bromine atoms. The bromine atoms initiate a chain reaction by their addition to the double bond of CF2CFBr.
The formation of CF2BrC(O)F as a principal product indicates that Br• atoms add to the CF2 group of the alkene. We have found no published data on the reaction rate constants for the addition of bromine atoms to the double bond of fluorinated olefins. As in the presence of CF2CFBr, bromine did not form, then the reaction (5) under the conditions of our work, must be more rapid, than the reaction (18) [42]. It indicates that the addition of Br• atoms to the fluorinated alkenes is faster that their reaction with chlorinated olefins. The reported room temperature rate constants for reactions of Br• with CHClCH2 [43] and CH2CCl2 [43] are (4.4 ±1.1) x 108 and (2.2±0.4) x 108 dm3 mol-1 s-1, respectively, those with CHClCCl2 are (6.6±2.4) x 107 [24,25], 9.4 x 107 [26] and (5.4±1.1) x 107 dm3 mol-1 s-1 [43] and those with CCl2CCl2 are (5.4±01) x 104 [25],< 7 x104 [26] and < 6 x106 dm3 mol-1 s-1 [43]. The room temperature rate constant for the reaction of Br• with CH2CH2 is 7.4 x 107 dm3 mol-1 s-1 [26]. The addition of chlorine atoms to chloroethenes at 298 K is fast, of order of 1010 dm3 mol-1 s-1[44].
Only very small amounts of the termination product, CF2BrCFBrNO2, was formed through reaction (16), corroborating the almost complete elimination of CF2BrCFBr• radicals by O2. The equilibrium studies of CCl3• + O2 ↔ CCl3O2• [45] and of R• + O2 ↔ RO2• [46-48], where R• are alkyl radicals, suggest that the elimination of radicals CF2BrCFBr• should be almost complete at the oxygen pressure used in our work. It was reported by other authors, that the fraction of ethyl radicals that escape oxidation is <0.1% at 2 torr of O2 [49].
Under conditions of our work some peroxy radicals react with NO2 to give CF2BrCFBrO2NO2. The calculated rate constants for CX3O2• + NO2 → CX3O2NO2, where X = H, F, Cl, are of order of 109 dm3 mol-1 s-1 [36] and that reported for CF3CFClO2• + NO2 + M→ CF3CFClO2NO2 + M is (3.5±0.3) x 109 dm3 mol-1 s-1 [50]. The formation of small amounts of trifluorobromoethene epoxide, indicates that a few radicals CF2BrCFBrO2• add to the double bond of alkene, reactions (8) and (9), regenerating CF2BrCFBrO•. The epoxidation of alkenes by addition of peroxyl radicals to the double bond, producing RO• radical and epoxide was reported [51]. The expression for ka, the rate constant for the addition of peroxy radicals RO2• to CF2CCl2, obtained in our previous work [21], is ka = (1.9±1) x 108 exp(-4.8±1.4 kcal mol-1 /RT) dm3 mol-1 s-1.
The formation of CF2BrC(O)F as the major product, indicates that the main via of disappearance of peroxyradicals CF2BrCFBrO2• is reaction (7) producing oxyradicals CF2BrCFBrO•. The rate constant for the reaction RO2• + RO2• → 2 RO• + O2 are of order of 109 dm3 mol-1 s-1. It was reported that rate constant for the self-reaction of CF3CFCO2• to give CF3CFCO• is (1.57±0.3) x 109 dm3 mol-1 s-1 [50].
The predominant fate of perhalogenated RO• is the halogen atom detachment, with C-C bond cleavage playing only a minor role. The elimination of Br• from the radical CF2BrCFBrO• can be explained in terms of concomitant weakening of the C-Br bond, when C-O bond is forming and the lower bond dissociation energy of the C-Br bond (~70 kcal/mol) as compared with the C-F bond (~110 kcal/mol). The extrusion of fluorine atom from perhalofluoromethoxy radicals is not expected as this process is highly endothermic [52]. The elimination of Br• from CF2BrCFBrO• predominates over C-C bond cleavage. The bond scission produces C(O)FBr and C(O)F2 and bromine atoms through the reaction sequence (11)-(14).
The pre-exponential factors, A, and activation energies, E, obtained for the decomposition of radicals CF2ClCCl2O• [21], CHCl2CCl2O• [22] and CCl3CCl2O• [23] were A = (7±3) x 1013 s-1 and E = 9.4±1.4 kcal mol-1 , A = (1.1±1) x 1014 s-1 and E = 9.4±2.4 kcal mol-1, and A = (3.0±1.4) x 1013 s-1 and E = 9.7±1 kcal mol-1, respectively. In the theoretical study of the decomposition of halogenated alkoxy radicals [53] the following kinetic parameters were calculated for temperature range 240-260 K: A = (4.06-4.49) x 1013 s-1 and E = 9.7 kcal/mol for CCl3O•, A = (2.85-3.1) x 1013 s-1 and E = 10.6 kcal mol-1 for CFCl2O•, and A = (2.85- 3.13) x 1013 s-1and E = 12.4 kcal mol-1 for CF2ClO•.
Molecular bromine is produced through reactions (17), (7), (8), (18) and the fast reaction (19), when all the alkene is consumed. Evidence was reported that the magnitude of the rate constant, k19, falls between 109 and 1010 dm3 mol-1 s-1 [42].
As the rate constant k17 = (2.2±1) x 10-5 s-1, for the decomposition of CF2BrCFBrO2NO2, is slow in comparison with those for the chain reaction steps, then in the time interval in which the alkene is present, the reaction (15) may be taken as a chain termination step together with the reaction (16). The reported room temperature rate constants for decomposition of CCl3O2NO2[54], CCl2FO2NO2 [54], CClF2O2NO2 [54], CF3O2NO2 [55], CCl3C(O) O2NO2 [54], CF3O2C(O)NO2 [54], CF2ClCCl2O2NO2 [16] and CH3C(O)O2NO2, PAN [56] are 0.19, 6.6 x 10-2, 4.0 x 10-2, 6.1 x 10-2, 1.2 x 10-4, 8.0 x 10-5, 1.3 x 10-4 and 3.2 x 10-4 s-1, respectively.
Applying the steady-state approximation to the mechanism in presence of alkene, the following expressions for the respective consumption of CF2CFBr and NO2 were obtained:
| -d[CF2CFBr]/dt = k1[NO2][ CF2CFBr] {1 + (k5/k1)[Br•]/[NO2] + (k8/k1)[CF2BrCFBrO2•]/[NO2]} | I |
| -d[NO2]/dt = k1[NO2][CF2CFBr]{1 + (k15/k1) [CF2BrCFBrO2•] [M]/ [CF2CFBr] + (k16/ k1)[CF2BrCFBr•][M] / [CF2CFBr]} | II |
Neglecting the contribution of the reactions of (4), (8) and (9) and assuming that [CF2BrCFBr•] ≈ [CF2BrCFBrO2•], as practically all the radicals CF2BrCFBr• react with O2 to give peroxyradicals CF2BrCFBrO2•, the following expression was deduced:
| k5 [Br•][[CF2CFBr] = (k10 + k11) [CF2BrCFBrO•] = k6 [CF2BrCFBr•][O2][M] {1 - (k15/k6) [NO2]/[O2]} | III |
At the pressures of oxygen used in this work, O2 has a third body character, as the reaction is of pseudo-zero order with respect to O2 as reactant.
The chain velocity depends on the generation rate of the radicals CF2BrCFBrO• by reactions (5)-(7). Each oxy radical releases one bromine atom through reactions sequence (10)-(14), reforming the CF2BrCFBr• radicals. Decreasing the pressure of NO2, increases the generation of the chain carriers CF2BrCFBrO2• and CF2BrCFBrO•. With increasing generation of the radicals CF2BrCFBrO2• the rate of the reaction (7) raises more rapidly that the rates of the reactions (8) and (15), favoring the formation of CF2BrC(O)F and the release of the bromine atom.
Conclusion
This investigation provides evidence, not reported until now, that the free bromine atoms can be released in the absence of light as a consequence of oxidation of CF2CFBr initiated by addition of NO2 to the double bond of trifluorobromoethene in the presence of molecular oxygen. The formation of bromodifluoroacetyl fluoride CF2BrC(O)F indicates that the bromine atoms add to the CF2 group of the alkene, forming radicals CF2BrCFBrO2• and CF2BrCFBrO• in the presence of oxygen. There are no data on the addition of Br• atoms to the fluorinated ethenes. The lack of formation of bromine in presence CF2CFBr indicates that the addition of Br• atoms to the double bond competes successfully with the fast reactions Br• + NO2 → BrNO2 and Br•+ BrNO2 → Br2 + NO2 and suggests that the reactions of bromine atoms with fluorinated alkenes are more rapid than those with the chlorinated counterpart. A new and simple way of preparation of CF2BrC(O)F is reported here. Previously it was prepared by catalytic reduction by lithium aluminum hydride of bromodifluoroacetate, obtained treating 1,2-dibromo-1- chlorotrifluoroethane with fuming H2SO4 and HgO [57]. The room temperature rate constant, k17 = (2.2±1) x 10-5 s-1 for the decomposition of peroxynitrate, CF2BrCFBrO2NO2 is presented for the first time.
Acknowledgment
The authors thank Mr. Z. Czarnowski for helpful comments. This work was supported by CONICET (National Science Research Council), Argentina. J.C. thanks financial support by the Max Planck Institute for Biophysical Chemistry Goettingen through the Partner Group for Chlorofluorocarbons in the Atmosphere (C. J. Cobos grant). R.M.R. thanks Fundación Antorchas (14022-3), CONICET (PEI-6177) and Volkswagen Foundation (Professors Oberhammer and Della Védova grant) for the purchase of the FTIR instrument.
References
[1] Dyatkin, B.L.; Mochalina, E.P.; Knunyants, I.L., Russian Chem. Rev., 1966, 35, 417. [ Links ]
[2] Barr, D.A.; Haszeldine, R.N., J. Chem. Soc., 1960, 1151. [ Links ]
[3] Fokin, A.V.; Uzun, A.T., Zh. Obshchei Khimii, 1966, 36, 117. [ Links ]
[4] Coffman, D.D.; Raasch, G.W.; Rigby, G.W.; Barrick, P.L; Hanford, W.E., J. Org. Chem., 1949, 14, 747. [ Links ]
[5] Haszeldine, R.N., J. Chem. Soc., 1953, 2075. [ Links ]
[6] Bissel, E., J. Org. Chem., 1961, 26, 5100. [ Links ]
[7] Knunyants, I.L.; Fokin, A.V.; Komarov, V.A., Zh. Vses. Khim. Obshchestvam. D. I. Mendeleeva, 1962, 7, 709. [ Links ]
[8] Knunyants, I.L.; Fokin, A.V.; Komarov, V.A., Izv. Akad. Nauk SSSR, Ser. Khim., 1966, 466. [ Links ]
[9] Spicer, Ch.W.; Heicklen, J., Int. J. of Chem. Kinet, 1972, 4, 575. [ Links ]
[10] Romano, R.M.; Della Védova, C.O.; Czarnowski, J.,Z. Phys. Chem., 2002, 216,1203. [ Links ]
[11] Czarnowski, J.; Schumacher, H.J., Int. J. Chem. Kinet., 1986, 18, 907. [ Links ]
[12] Czarnowski, J.,Int. J. Chem. Kinet., 1992, 24, 679. [ Links ]
[13] Romano, R.M.; Czarnowski, J., Z. Phys. Chem., 2004, 218, 575. [ Links ]
[14] Romano, R.M.; Czarnowski, J., Z. Phys. Chem., 2005, 219, 849. [ Links ]
[15] Czarnowski, J., J. Chem. Soc. Perkins Trans. 2, 1991, 1459. [ Links ]
[16] Czarnowski, J., Bull. Polish Acad. Science Chem., 1991, 39, 49. [ Links ]
[17] Niki, H.; Maker, P.D; Savage, C.M.; Preitenbach, L.P.; Hurley, M.D., Int. J. Chem. Kinet., 1986, 18, 1235. [ Links ]
[18] Czarnowski, J.; Castellano, E.; Schumacher, H.J.Z, Phys. Chem. NF, 1969, 65, 225. [ Links ]
[19] Czarnowski, J.; Schumacher, H.J.Z., Z. Phys. Chem. NF, 1970, 73, 68-76. [ Links ]
[20] Kennedy, R.C.; Levy, J.B., J. Phys. Chem., 1972, 76, 3480. [ Links ]
[21] Czarnowski, J.,J. Chem. Soc. Faraday Trans., 1989, 85, 1425. [ Links ]
[22] Czarnowski, J., Z. Phys. Chem., 1995, 191, 103, [ Links ]
[23] Czarnowski, J., Z. Phys. Chem., 1998, 203, 183 [ Links ]
[24] Catoire, V.; Ariya, P.A.; Niki, H.; Harris, G.W., Int. J. Chem. Kinet., 1997, 29, 695. [ Links ]
[25] Ariya, P.A.; Catoire, V.; Sander, R.; Niki, H.; Harris, G.W., Tellus, 1997, 49B, 583. [ Links ]
[26] Ramacher, B.; Orlando, J.J., G. S. Tyndall, Int. J. Chem. Kinet., 2001, 33, 198. [ Links ]
[27] Park, J.D.; Snow, C.M.; Lecher, J.R., J. Am. Chem. Soc., 1951, 73, 2342. [ Links ]
[28] Nielsen, J.R.; Richard, C.M.; Mc Murry, H.L., J. Chem. Phys., 1948, 16, 67. [ Links ]
[29] Arce, V.; Czarnowski, J.; Romano, R.M., to be published. [ Links ]
[30] Drew, B.R.; Gounev, T.K.; Guirguis, G.A.; Durig, J.B., J. Raman Spectrosc., 1998, 29, 205. [ Links ]
[31] Nielsen, A.H.; Burke, T.G.; Woltz, P.J.W., E. A. Jones, J. Phys. Chem., 1952, 20, 596. [ Links ]
[32] Parkington, M.J.; Ryan, T.A.; Seddon, K.R., J. Chem. Soc. Dalton Trans., 1997, 251. [ Links ]
[33] García, P.; Willner, H.; Oberhammer, H.; Francisco, J.S., J. Chem. Phys., 2004, 121, 11900. [ Links ]
[34] Niki, H.; Maker, P.D.; Savage, C.M.; Breitenbach, L.P., Chem. Phys. Lett., 1979, 61, 100. [ Links ]
[35] Köppenkastrop, D.; Zabel, F., Int. J. Chem. Kinet., 1991, 23, 1. [ Links ]
[36] Forst, W.; Caralp, F., J. Phys. Chem., 1992, 96, 6291. [ Links ]
[37] Kopitzky, R.; Willner, H.; Mack, H.; Pfeiffer, A.; Oberhammer, H., Inorg. Chem., 1998, 37, 6208. [ Links ]
[38] Zabel, F.; Kirchner, F.; Becker, K.H., Int. J. Chem. Kinet., 1994, 26, 827. [ Links ]
[39] Chow, D.; Jones, M.H.; Thorne, M.P.; Wong, E.C., Can. J. Chem., 1969, 47, 2491. [ Links ]
[40] Hubinger, S.; Nee, J.B., J. Photochem. Photobiol., A: Chem., 1995, 86, 1. [ Links ]
[41] Blend, H., J. Chem. Phys., 1970, 53, 4497 [ Links ]
[42] Orlando, J.J.; Burkholder, J., J. Phys. Chem., 2000, 104, 2048. [ Links ]
[43] Bierbach, A.; Barnes, I.; Becker, K.H., Int. J. Chem. Kinet., 1996, 28, 565. [ Links ]
[44] Atkinson, R.; Aschmann, S.M., Int. J.Chem. Kinet., 1987, 19, 1097. [ Links ]
[45] Russel, J.J.; Seetula, J.A.; Gutman, D.; Danis, F.; Caralp, F.; Lightfoot, P.D.; Lesclaux, R.; Melius, C.F.; Senkan, S.M., J. Phys. Chem., 1990, 94, 3277. [ Links ]
[46] Slage, I.R.; Ratajczak, E.; Heaven, M.C.; Gutman, D.; Wagner, A.F., J. Am. Chem. Soc., 1985, 107, 1838. [ Links ]
[47] Slage, I.R.; Gutman, D., J. Am. Chem. Soc., 1985, 107, 5342. [ Links ]
[48] Slage, I.R.; Ratajczak, E.; Gutman, D., J. Phys. Chem., 1986, 90, 402. [ Links ]
[49] Adachi, H.; Basco, N.; James, D.G.L., Int. J. Chem. Kinet., 1979, 11, 121. [ Links ]
[50] Sehested, J., Int. J. Chem. Kinet., 1994, 26, 1023. [ Links ]
[51] Stark, M.S., J. Phys. Chem., 1997, 101, 8296. [ Links ]
[52] Rayez, J.C.; Rayez, M.T.; Halvick, P.; Duguay, B.; Danneberg, J.J., Chem. Phys., 1987, 118, 265. [ Links ]
[53] Rayez, J.C.; Rayez, M.T.; Halvick, P.; Duguay, B.; Lesclaux, R.; J. J. Danneberg, J.J.,Chem. Phys., 1987, 116, 203. [ Links ]
[54] Zabel, F., Z. Phys. Chem., 1995, 188, 119. [ Links ]
[55] Mayer-Figge, A.; Zabel, F.; Becker, K.H., J. Phys. Chem., 1996, 100, 6587. [ Links ]
[56] Roberts, J.M.; Bertman, S.B., Int. J. Chem. Kinet., 1992, 24, 297. [ Links ]
[57] Cambell, R.W.; Vogl, O., Die Macrom. Chem., 1979, 180, 633. [ Links ]

