




![Two isostructural complexes of Co(II) and Zn(II) with lapacholate, dimethylformamide and water, [M(Lap)2(DMF)(H2O)]](/img/es/next.gif)





Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Asociación Química Argentina
versión impresa ISSN 0365-0375
An. Asoc. Quím. Argent. v.93 n.4-6 Buenos Aires ene./dic. 2005
REGULAR PAPERS
Arrhenius parameters for the thermal decomposition of FC(O)OOC(O)F
Burgos Paci, M.A.; Argüello, G.A
INFIQC. Departamento de Físico Química, Facultad de Ciencias Químicas, Universidad, Nacional de Córdoba. Ciudad Universitaria, 5000 Córdoba, Argentina, Fax: +54 351 433 4188, E-mail: gaac@fisquim.fcq.unc.edu.ar
García, P., Pernice H., Willner, H.
Fachbereich C, Anorganische Chemie, Bergische Universität Wuppertal, D-42119 Wuppertal, Germany
Received August 23th, 2005. In final form September 30th, 2005
Dedicated to the memory of the late Prof. Hans J. Schumacher on the occasion of his 100th birthday
Abstract
The gas phase thermal decomposition of FC(O)OOC(O)F was studied at temperatures between 40 and 70 ºC using FTIR spectroscopy to follow the course of the reaction. The decomposition was studied at different partial pressures of NO between 90 to 180 mbar and using N2 to set the total pressure to 1000 mbar. The rate constant fits the Arrhenius equations ![]() from which the O—O bond energy of (23.9 ± 0.9) kcal mol-1 was obtained.
from which the O—O bond energy of (23.9 ± 0.9) kcal mol-1 was obtained.
Resumen
La descomposición térmica del FC(O)OOC(O)F fue estudiada entre 40 y 70 ºC por medio de espectroscopía FTIR para seguir el curso de la reacción. La descomposición se estudió a diferentes presiones de NO (90 a 180 mbar) agregando N2 para dar una presión final de 1000 mbar. La constante de descomposición se ajusta a la ecuación ![]() a partir de la cual se determina la energía de enlace O—O como (23,9 ± 0,9) kcal mol-1.
a partir de la cual se determina la energía de enlace O—O como (23,9 ± 0,9) kcal mol-1.
Introduction
Since the first synthesis of FC(O)OOC(O)F by Schumacher et al.[1] more than forty five years ago, much work has been done with this and other related halogen containing molecules. The ideas prevailing in the sixties, and particularly in Schumacher´s group, were at the time logically away from the urgencies of today's atmospheric sciences, though they were involved in countless projects dealing with halogenated molecules that later proved to have substantial impact on the chemistry of the terrestrial atmosphere [2-4]. After the recognition of the deleterious effect that the chlorofluorocarbons (CFCs) had on the ozone layer [5,6] and their replacement by hydrofluorocarbons, (HFCs) there have been exhaustive studies of the mechanisms, intermediates and final products involved in many of these halogenated molecules. In the last decade much work has been devoted to the study of the properties and reactions of many compounds and radicals containing only F, C and O atoms that can be formed in the laboratory as a result of the degradation of HFCs in the presence of oxygen and high concentrations of CO. The study of these reactions afforded many new compounds to be synthesized and used as precursors of atmospherically relevant radicals which were thus isolated [7-9]. One of such compounds is precisely FC(O)OOC(O)F that besides having being synthetized in more than one way [1,10- 12], was used as a polymerization initiator [13]. Further studies on this peroxide over four decades involved the elucidation of its IR spectrum [14,15], studies of its gas phase structure [16], its chemistry [17] and its potential as reagent for synthesis [18,19]; a theoretical density functional study [20] and its involvement in atmospheric chemistry as a precursor of FCO2 radicals that were isolated in low temperature matrices [21] and studied in their reactions with other atmospheric gas traces [22].
Kinetic data on thermal decomposition are needed in order to have reliable estimates of bond energies and to help in the elucidation of mechanisms and in the calculation of thermodynamic properties. Nevertheless, for fluorocarbonoxygenated peroxides and trioxides this kind of studies have been reported for just a few compounds like CF3OOCF3 [23], CF3OOOCF3 [24], CF3OC(O)OOC(O)OCF3 [25], CF3OC(O)OOC(O)F and CF3OC(O)OOCF3 [26]. For the title compound, Schumacher's group gave a value of the O-O bond energy, derived from indirect studies [13]. In the present work the thermal decomposition rate constants for FC(O)OOC(O)F have been measured as a function of temperature and in the first order region providing a direct determination of the bond energy. The Arrhenius parameters obtained are discussed in comparison to the former bond energy determination and conform a contribution to the available atmospheric databases.
Results
The thermal decomposition of FC(O)OOC(O)F in presence of NO was evaluated at temperatures between 40 and 70 °C, using N2 to set a total pressure of 1000 mbar. The disappearance of the reagent was followed using its absorption band at 1185 cm-1 without need of subtraction from interfering products. The data were analyzed according to first-order kinetics:
|
| (1) |

The first step in the decomposition of FC(O)OOC(O)F is the formation of FCO2 radicals, which have a half-life long enough to recombine to give back the peroxide. A way to obtain the correct values for the kinetic parameters of the decomposition reaction is the rapid reaction of FCO2 radicals by the addition of an effective scavenger. We studied the decomposition reaction of FC(O)OOC(O)F in presence of NO because it is well known that it reacts efficiently with FCO2 radicals [27]. The partial pressure of NO was varied between 90 and 180 mbar at each temperature of decomposition.
Figure 1 shows plotting of the logarithms of absorbance vs. time for reactant loss with 120 mbar of NO. Good straight lines (not shown in the figure) were obtained for all NO pressures. At each temperature studied, the first-order rate constant was calculated by a least-squares method. Average rate constants are given in Table 1.
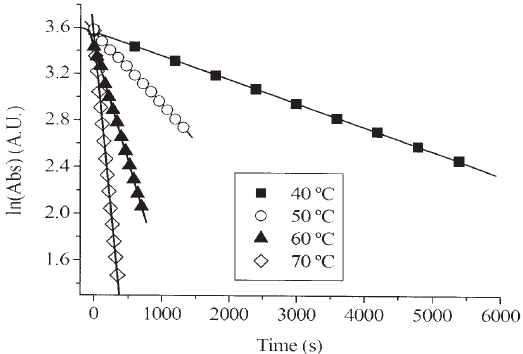
Figure 1: First-order kinetic curves at selected temperatures for the decomposition of FC(O)OOC(O)F.
Table1. First-order rate constants from decomposition of FC(O)OOC(O)F in NO at different temperatures

To evaluate the Arrhenius parameters, the determined rate constants were used to construct an Arrhenius plot for every NO partial pressure, obtaining very good straight lines from the linear least-squares analysis in each case. Nevertheless, since the differences in the Arrhenius parameters for the different partial pressures of NO are within the experimental uncertainties, the Arrhenius equation for the decomposition was fitted to all the experimental points as can be seen in Figure 2. In this manner, only one averaged value was determined as:
|
| (2) |

Figure 2: Arrhenius plot for the decomposition of FC(O)OOC(O)F.
Figure 3 shows the IR spectra of reactants and/or products at different times of reaction. It can bee seen that the decomposition results in the formation of CO2 (2300 cm-1), HF (3830-4140 cm-1), NO2 (1600 cm-1) and SiF4 (1022 cm-1). In some experiments FNO (1843 cm-1) was detected as a transient species.

Figure 3: IR spectra of the reactants (lower trace) and products obtained (upper trace) in the decomposition of FC(O)OOC(O)F.
Discussion
The first step in the decomposition process of FC(O)OOC(O)F should be the breaking of the peroxide bond, the weakest bond in the molecule. The O—O rupture produces two FCO2 radicals
| FC(O)OOC(O)F → 2 FCO2 | (3) |
In presence of NO, these FCO2 radicals will react to form FNO and CO2
| FCO2 + NO → FNO + CO2 | (4) |
Slightly differing values (3.6-3.9 x10-11cm3 s-1) of the rate coefficient for this reaction have been measured [27-29] at room temperature. Comparing these values with the rate coefficient for the recombination of FCO2 radicals to give the peroxide back, 7x10-13 cm3 s-1, it is clear that the back reaction (-3) is absolutely suppressed [28-30].
The FNO was detected as a transient product in some experiments. After its formation via reaction (4), heterogeneous reactions with the walls of the reaction cell could have been responsible of the NO2 formation, as well as of the appearance of HF and SiF4.
Arvia et al. [11] studied the reaction of FC(O)OOC(O)F with NO at room temperature, they explained their results in terms of the overall reaction
| FC(O)OOC(O)F + NO → NO2 + CF2O + CO2 | (5) |
Though there was no detailed indication of the amount of NO, the authors indicate that the reaction between the peroxide and NO was carried out in excess NO. As stated before, we found NO2 and CO2 as reaction products, but we did not observe the formation of CF2O. In our experiments the partial pressure for the peroxide was 1.5-2.2 mbar and always at least 90 mbar of NO were added. Under these experimental conditions all the FCO2 radicals should have reacted with NO through reaction (4).
FC(O)OOC(O)F decomposition in the presence of NO was studied at total pressures of 1000 mbar set with N2. It is assumed that under these conditions the reaction runs in the high pressure region, as expected for a molecule of this complexity. Examples of similar peroxide species that achieve their first-order values at pressures higher than 500 mbar are (CF3OC(O)O)2 [25], (CH3C(O)O)2 [31], CF3OOOCF3 [24] and CH3C(O)OONO2 [32].
The derived activation energy is 23.9 kcal/mol and the extrapolated A factor is 1.0x1013. The A factor obtained can be compared with those collected in Table 2. It can be seen that it is the lowest of the peroxides listed, and therefore indications pointing to some complex mechanism could be advanced. Nevertheless, we do believe the decomposition directly provides two smaller fragments in a simple bond fission. This hypothesis has been certified through the isolation of the FCO2 radicals in low temperature matrices [21], prepared from pyrolysis of FC(O)OOC(O)F. Therefore, the difference in A factors should be rationalized in other terms. Also for comparison it is included in the Table the trioxide CF3OC(O)OOOC(O)OCF3 for which an even lower A factor has been reported. The pyrolysis of this molecule afforded, again in a simple direct decomposition reaction, the primary radicals that were subsequently trapped in low temperature matrices [8]. Therefore, simple direct O-O bond rupture can occur with A factors usually encountered for more complex reactions.
Table 2. Pre-exponential factors and entropy of activation for selected peroxides.

In terms of the π stabilization afforded by the two fragments formed, it can be seen from the Table that the hydrogenated peroxides have almost the same A factor (ranging from 1014.2 to 1014.4) as the fluorinated peroxide CF3OC(O)OOC(O)OCF3 (with an A factor of 1014.5). All these peroxides form two equal, bulky and delocalized acyloxy-radicals when decomposed. When a smaller fragment takes part in the molecule there is no simple correlation within the A factors. In the case of CF3OC(O)OOC(O)F, again two contributions to π stabilization are possible (one through the FCO2 radical [21] and the other through CF3OCO2) and the A factor is the highest.
The distinct feature shown in the Table is the activation entropy. FC(O)OOC(O)F is the only peroxide having a negative value. This could be interpreted not only in terms of forming two smaller fragments in the dissociation but also in acquiring the proper orientations for the leaving fragments during the decomposition.
A general overview of the bond energy for the peroxides shows that it increases when the peroxydic bond is directly attached to a non-carbonylic group having large electron withdrawing capacity.
The bond energy for FC(O)OOC(O)F has been the subject of four independent determinations. Arvia et al. [13] have informed a value of 31 ± 3 kcal mol-1 though their determination was not a direct one. Years later, McKee et al. [20] performed a theoretical calculation using the most refined numerical tools available at that time. Their value of 14.8 kcal mol-1 is far away from the numbers in Table 2 as is also the value of Cobos et al. [33] of 30.9 kcal mol-1 that was derived theoretically from isodesmic reactions. Our value is the only one that comes from a direct determination and falls around the middle. A lower value for the dissociation energy than the more frequent 30 kcal mol-1 seems reasonable in the light of the stability of the radicals formed, since FCO2 radicals are very stable and long lived. Also reasonable seems the 34 kcal mol-1 for the peroxide CF3OC(O)OOCF3 that does not have one of the two C(O) blocking units that protect the withdrawing of non-bonding electrons from the peroxidic bond.
The theoretical value of 14.8 kcal mol-1 calculated at the B3LYP/6-31+G(d) level of theory by McKee et al. is unreasonable low in terms of the stability of the peroxide; since with this number as activation energy and the pre-exponential factor measured in this work, the half-life for the thermal decomposition at room temperature should be around 1 ms. The problems associated with the calculation of bond fission energies as the direct difference of the energy of products and reactants for the dissociation reaction are well known and Cobos et al. have discussed the case for FC(O)OOC(O)F in Ref 33. The theoretical value of 30.9 kcal mol-1 obtained by Cobos et al. from isodesmic reactions is higher than ours in around seven kcal mol-1. The use of isodesmic reactions is a very useful method for the evaluation of heats of formation with relative errors between 2-5%. Assuming the errors for the heats of formation of FC(O)OOC(O)F and FCO2 as ±4 and ±2 kcal mol-1 respectively, the energy for the O-O bond fission reaction would give 31±5. A comparison with our value of 23.9 kcal mol-1 still gives a difference beyond the error limits, but the discrepancy is rather small.
Experimental Section
All volatile materials were manipulated in a glass vacuum line equipped with two capacitance pressure gauges (Bell and Howell and MKS Baratron 220) and three U-traps connected via glass valves with PTFE stopcocks (Young, London). The vacuum line was connected to a photoreactor, a thermal reactor and to a double-walled IR gas cell (optical path length 20 cm, Si windows 0.5 mm thick) placed in the sample compartment of an FTIR spectrometer (Bruker IFS28). This arrangement made it possible to follow either the course of the synthesis, the purification processes or the thermal decay of substances.
The synthesis of the FC(O)OOC(O)F was carried out following the original Schumacher's recipe [1], which is the best method so far.
The thermal decomposition of FC(O)OOC(O)F was studied using a double-walled IR cell in the sample compartment of the spectrometer. The outer jacket of the cell was connected to a thermostat, from which hot salty water flowed at temperatures ranging between 40 and 70 °C with an uncertainty of ± 0.1 °C. Once the cell reached the intended temperature, 90-180 mbar NO were admitted and then a diluted mixture of peroxide in N2 was introduced. This procedure ensures the mixing of the reactants in a short time. The partial pressures of the peroxide in the cell ranged between 1.5 - 2 mbar and the total pressure was 1000 mbar. After the reaction was started a series of in-situ timely spaced IR spectra were obtained. The data processing of the kinetic measurements was done integrating the absorption band at 1185 cm-1 which corresponds to the strongest band of the peroxide. In this manner it is possible to follow the disappearance of the peroxide without looking to the possible heterogeneous reactions of the FCO2 radicals that very probably occurred in Schumacher's experiments since they could only look at the total pressure modification.
The products obtained (CO2, FNO, SiF4 and HF) were identified from reference spectra of pure samples.
Acknowledgment
We gratefully acknowledge financial support from VW Stifftung, FONCyT and CONICET. MABP wishes to thank CONICET for his post-doctoral fellowship. PG wants to thank the PROALAR project for covering his travel expenses to Argentina. Language assistance from Karina Plasencia is gratefully acknowledged.
References
[1] Arvia, A. J.; Aymonino, P. J.; Waldow, C. H.; Schumacher, H. J. Angew. Chem. 1960, 72, 169. [ Links ]
[2] Cafferata, L. F. R.; Sicre, J. E.; Schumacher, H. J. Z. Phys. Chem. 1961, 29, 188. [ Links ]
[3] Staricco, E. H.; Sicre, J. E.; Schumacher, H. J. Z. Phys. Chem. 1962, 31, 385. [ Links ]
[4] Viscido, L.; Sicre, J. E.; Schumacher, H. J. Z. Phys. Chem. 1962, 32, 182. [ Links ]
[5] Montzka, S. A.; Butler, L. J.; Elkins, J. W.; Thompson, T. M. A.; Clarke, D.; Lock, L. T. Nature. 1999, 398, 690. [ Links ]
[6] Sekiya, A.; Misaki, S.; Beppu, T. The Earth Technologies Forum. The conference on climate change and Ozone Protection. 2003. [ Links ]
[7] Argüello, G. A.; Willner, H. J. Phys. Chem. A. 2001, 105, 3466. [ Links ]
[8] von Ahsen, S.; Willner, H.; Francisco, J. S. Chem. Eur. J. 2002, 8, 4675. [ Links ]
[9] von Ahsen, S.; Hufen, J.; Willner, H.; Francisco, J. S. Chem. Eur. J. 2002, 8, 1189. [ Links ]
[10] Arvia, A. J.; Aymonino, P. J.; Schumacher, H. J. Anal. Asoc. Quím. Arg. (This Journal) 1962, 50, 135. [ Links ]
[11] Arvia, A.J.; Aymonino, P. J.; Schumacher, H. J. Z. Anorg. Allgem. Chem. 1962, 316, 327. [ Links ]
[12] Czerepinski, R.; Cady, G. H. Inorg. Chem. 1968, 7, 169. [ Links ]
[13] Arvia, A.J .; Aymonino, P. J.; Schumacher, H. J. Z. Phys. Chem. 1961, 28, 393. [ Links ]
[14] Arvia, A. J.; Aymonino, P. J. Spectrosc. Acta. 1963, 18, 1299. [ Links ]
[15] Della Vedova, C. O.; Mack, H. G. J. Molec. Struc. 1992, 274, 25. [ Links ]
[16] Mack, H.-G.; Della Vedova, C. O.; Oberhammer, H. Angew. Chem. 1991, 103, 1166. [ Links ]
[17] Fox, W. B.; Franz, G. Inorg. Chem. 1966, 5, 946. [ Links ]
[18] Pilipovich, D.; Schack, C. J.; Wilson, R. D. Inorg. Chem. 1972, 11, 2531. [ Links ]
[19] Talbott, R. L. J. Org. Chem. 1968, 33, 2095. [ Links ]
[20] McKee, M. L.; Webb, T. R. J. Phys. Chem. 1996, 100, 11292. [ Links ]
[21] Argüello, G. A.; Grothe, H.; Kronberg, M.; Willner, H.; Mack, H.-G. J. Phys. Chem. 1995, 99, 17525. [ Links ]
[22] Bednarek, G.; Argüello, G. A.; Zellner, R. Ber. Bunsenges. Phys. Chem. 1996, 100, 445. [ Links ]
[23] Kennedy, R. C.; Levy, J. B. J. Phys. Chem. 1972, 76, 3480. [ Links ]
[24] Czarnowski, J.; Schumacher, H. J. Int. J. Chem. Kinet. 1981, 13, 639. [ Links ]
[25] Burgos Paci, M. A.; Argüello, G. A.; García, P.; Willner, H. Int. J. Chem. Kinet. 2003, 35, 15. [ Links ]
[26] Burgos Paci, M. A.; Argüello, G. A.; García, P.; Willner, H. J. Phys. Chem. A. 2005, 109(33), 7481. [ Links ]
[27] Mörs, V.; Argüello, G. A.; Hoffmann, A.; Malms, W.; Röth, E. P., Zellner, R. J. Phys. Chem. 1995, 99, 15899. [ Links ]
[28] Wallington, T. J.; Ellermann, T.; Nielsen, O. J.; Sehested, J. J. Phys. Chem. 1994, 98, 2346. [ Links ]
[29] Maricq, M. M.; Szente, J. J.; Dibble, T. S.; Francisco, J. S. J. Phys. Chem. 1994, 98, 12294. [ Links ]
[30] Croce, A. E.; Cobos, C. J.; Castellano, E. Chem. Phys. 1996, 211, 215. [ Links ]
[31] Rembaum, A.; Szwarc, M. J. Am. Chem. Soc. 1954, 76, 5975. [ Links ]
[32] Cox, R. A.; Roffey, M. J. Environ Sci. & Technol. 1977, 11, No 9, 900. [ Links ]
[33] Badenes, M. P.; Castellano, E.; Cobos, C. J.; Croce, A. E.; Tucceri, M. E. Chem. Phys. Lett. 1999, 303, 482. [ Links ]
[34] Argüello, G. A.; von Ahsen, S.; Willner, H.; Burgos Paci, M. A.; García, P. Chem. Eur. J. 2003, 9, 5135. [ Links ]
[35] Rembaum, A. Szwarc, M. J. Chem. Phys. 1955, 23, 909. [ Links ]

