



![Two isostructural complexes of Co(II) and Zn(II) with lapacholate, dimethylformamide and water, [M(Lap)2(DMF)(H2O)]](/img/es/prev.gif)






Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Asociación Química Argentina
versión impresa ISSN 0365-0375
An. Asoc. Quím. Argent. v.93 n.4-6 Buenos Aires ene./dic. 2005
REGULAR PAPERS
Salt (Electrolytes) Sorption By Membranes And/Or Ion-Exchange Resins: Donnan Or Partition Equilibrium?
Timmermann, E.O.
Departamento de Química, Facultad de Ingeniería, Universidad de Buenos Aires, Paseo Colón 850, (1305) Buenos Aires Fax: +54 4331 0129, Email: etimmer@fi.uba.ar
Received September13th, 2005. In final form December 6th, 2005
Dedicated to the memory of the late Prof. Hans J. Schumacher on the occasion of his 100th birthday
Abstract
The sorption equilibrium of salts (electrolytes) between aqueous solutions and ionexchanging phases (membranes, resins, and/or gels) is analysed. Classically this equilibrium had been treated as Donnan equilibrium with unitary constants (Teorell (1953), Schloegl (1964)). However, as phases of different structure and different thermodynamic standard states are involved a partition equilibrium with non-unitary constants is the correct thermodynamic description, as it is used, p.ex. in the solubility-diffusion theory of membrane processes. Surprisingly a complete thermodynamic description of such equilibrium has not been given. Stating the thermodynamic phase equilibrium conditions directly for the salts (not for ions) and taking into account the molarity (X) of fixed ions in the ion exchanging phase (accountable of the characteristic ion exclusion effect) a limiting sorption law for a binary salt of type M ν1Yν2 is obtained (indices: 1 = counter-ion, 2 = co-ion within the sorbing phase):
![]() ,
,
(qs = ν2 |z2| cs/X; cs = salt molarity in the membrane;![]() external molal salt activity) law which depends on the salt formula (relation ν1/ ν2) and the membrane characteristics. Experimental sorption data of different salt/membrane systems (NaBr/NaR; CsBr/CsR; SrBr2 /SrR; MgCl2 /RCl; MgCl2 /MgR; LaCl3 /LaR; Na2SO4 /NaR) follow this law. In log-log graphs of qs vs.
external molal salt activity) law which depends on the salt formula (relation ν1/ ν2) and the membrane characteristics. Experimental sorption data of different salt/membrane systems (NaBr/NaR; CsBr/CsR; SrBr2 /SrR; MgCl2 /RCl; MgCl2 /MgR; LaCl3 /LaR; Na2SO4 /NaR) follow this law. In log-log graphs of qs vs.![]() the experimental exponent ns coincides with its theoretical values (1+ ν1/ ν2). The partition constants are also obtained as well as the activity coefficient of the salt within the sorbing phase. It is found that this coefficient (>1) is logarithmically linear in √(Is), the ionic strength of the salt in this phase. Finally the complete sorption equations for qs are solved, i.e. eqs. of 2nd. degree (symmetric electrolytes) and of 3rd. degree (1:2 electrolytes), the latter being treated in detail. The expressions of the partition factors of the counter-ion and of the co-ion are also obtained as well as their limiting expressions.
the experimental exponent ns coincides with its theoretical values (1+ ν1/ ν2). The partition constants are also obtained as well as the activity coefficient of the salt within the sorbing phase. It is found that this coefficient (>1) is logarithmically linear in √(Is), the ionic strength of the salt in this phase. Finally the complete sorption equations for qs are solved, i.e. eqs. of 2nd. degree (symmetric electrolytes) and of 3rd. degree (1:2 electrolytes), the latter being treated in detail. The expressions of the partition factors of the counter-ion and of the co-ion are also obtained as well as their limiting expressions.
Resumen
Se analiza el equilibrio de sorción de sales (electrolitos) entre soluciones acuosas y fases intercambiadoras de iones (membranas, resinas, y/o geles). Clásicamente este equilibrio ha sido tratado como equilibrio Donnan con constantes unitarias (Teorell (1953), Schloegl (1964)). Sin embargo, ya que se tratan fases de distinta estructura y de distintos estados termodinámicos tipo un equilibrio de partición con constantes no-unitarias es la correcta descripción termodinámica, como es usada, p. ej., en la teoría de solubilidaddifusión para procesos de membrana. Sorprendentemente una descripción termodinámica completa de este equilibrio no se ha dado. Planteando las condiciones para el equilibrio termodinámico de fases directamente para sales (no para iones) y teniendo en cuenta la molaridad (X) de los iones fijos en la fase intercambiadora de iones (responsable del característico efecto de exclusión iónica) se obtiene una ley límite de sorción para sales binarias del tipo M ν1Y ν2 (índices: 1 = contraión, 2 = coión en la membrana):
![]() ,
,
(qs = ν2 |z2| cs/X; cs = molaridad salina en la membrana;![]() actividad salina molal externa) ley que depende de la fórmula de la sal (relación ν1/ ν2) y de las características de la membrana. Datos experimentales de sorción para diferentes sistemas de sales/membrana (NaBr/NaR; CsBr/CsR; SrBr2 /SrR; MgCl2 /RCl; MgCl2 /MgR; LaCl3 /LaR; Na2SO4 /NaR) siguen esta ley. En gráficos log-log de qs vs.
actividad salina molal externa) ley que depende de la fórmula de la sal (relación ν1/ ν2) y de las características de la membrana. Datos experimentales de sorción para diferentes sistemas de sales/membrana (NaBr/NaR; CsBr/CsR; SrBr2 /SrR; MgCl2 /RCl; MgCl2 /MgR; LaCl3 /LaR; Na2SO4 /NaR) siguen esta ley. En gráficos log-log de qs vs.![]() el exponente experimental ns coincide con su valor teórico (1+ ν1/ ν2) Se obtienen también las constantes de partición así como el coeficiente de actividad de la sal en la fase sorbente. Se encuentra que este coeficiente (>1) es logarítmicamente lineal en √(Is), la fuerza iónica de la sal en esa fase. Finalmente se resuelven las ecuaciones completas de sorción para qs, esto es, ecs. de 2do. grado (sales simétricas) y de 3er. grado (electrolitos 1:2), tratándose éstas últimas en detalle. Se obtienen además las expresiones para los factores de partición para contraión y coión así como sus expresiones límites.
el exponente experimental ns coincide con su valor teórico (1+ ν1/ ν2) Se obtienen también las constantes de partición así como el coeficiente de actividad de la sal en la fase sorbente. Se encuentra que este coeficiente (>1) es logarítmicamente lineal en √(Is), la fuerza iónica de la sal en esa fase. Finalmente se resuelven las ecuaciones completas de sorción para qs, esto es, ecs. de 2do. grado (sales simétricas) y de 3er. grado (electrolitos 1:2), tratándose éstas últimas en detalle. Se obtienen además las expresiones para los factores de partición para contraión y coión así como sus expresiones límites.
Introduction
The sorption equilibrium of an electrolyte between an aqueous solution and a membrane with fixed electrical charges or an ion-exchange resin has been described in different ways in the literature: (a) with an unitary equilibrium constant (Teorell (1953) [1], Schlögl (1964)[2], (b) with non-unitary constants (solubility-diffusion theory for membrane processes (1949) [3,4], but always considering that the phase-equilibrium solution-membrane is characterised by the so-called Donnan equilibrium.
In this paper, we revise these formulations through thermodynamics stating directly the phase equilibria in terms of the neutral components - the salts - involved and not by the usual ionic equilibria. Thermodynamically, only the quantities corresponding to neutral components are measurable, exhibiting concrete physical meaning. The effect of the fixed charges (X) of the membrane (common ion effect) is specifically taken into account, but not the electrical aspects because they compensate each other being not necessary to consider them explicitly. General laws, and their limiting expressions, are obtained and are applied to several experimental cases. The corresponding equilibrium constants are determined as well as the activity coefficients of the salts in the membrane phase. The analysis confirms our description, which is in some aspects differs and in others coincides and complements the findings of the literature [1-8].
Thermodynamic foundations
The Donnan equilibrium [9,10] describes the osmotic equilibrium between two aqueous electrolytic solutions separated by a membrane, one of the solutions containing a polyelectrolyte the macro-ion of which cannot permeate through the membrane. The presence of the macro-ion and its non-permeability through the membrane determines at equilibrium, on one side, an osmotic pressure difference between both solutions and, on the other side, an asymmetric ion distribution between the two solutions (essentially for the counter-ions of the macro-ion). Besides, this ionic asymmetry determines an electrical potential difference (Donnan potential) between both solutions.
It must be pointed out that, in the Donnan equilibrium formulation, only the thermodynamic properties of the components of the electrolytic solutions are involved, being all these welldefined [9,10]. The role of the membrane is then merely the one of a device preventing the access of the macro-ion to the other solution. This means that the membrane is completely passive (black box) being of no interest any of its other properties.
On the other hand, if we concentrate our attention to the two solution/membrane interfaces involved in the Donnan equilibrium the situation is different. To formulate thermodynamically these phase equilibria the characteristics of the membrane phase must be specifically stated. These interfaces are permeable to the salts and the solvent and the membrane substance now takes the role of the macro-ion if it possesses fixed electrical charges.
Consequently, differences arise from the fact that phases of different physical constitution are considered. In the membrane, the poly-ionic membrane substance is the major component, sorbing the substances (salts and solvent) of the external aqueous solution. Thus, one deals directly with a partition or distribution equilibrium of genuine nernstian type, while the fixed charges of the membrane determine the ion exclusion effect.
As usual, the equalities of the chemical potentials of the substances which partitionate or distribute through the interface between the two involved phases describe thermodynamically the equilibrium and the corresponding standard potentials in each phase must be carefully characterised. These standard potentials are identical for the Donnan Equilibrium as phases of identical physical constitution (the two external solutions) are considerated and, therefore, the corresponding equilibrium constants are unitary. But, if the involved phases are physically different as in the solution/membrane equilibrium, these standard potentials are different and correspondingly the partition constants become non-unitary. For very dilute external solutions, the membrane in equilibrium tends to the sorption equilibrium stage of pure water, constituting this state the thermodynamic reference state of infinite dilution of the salts in the membrane. It is the water saturated (swollen) state which is characteristic of each membrane system. Thereby, the nonunitary partition constant is specific for each solution/membrane equilibrium. This condition differs from the conventional Donnan equilibrium and establishes the basis of the present work. Moreover, its results are requisites for the analysis of the transport phenomena within the membrane in terms of phenomenological mobilities [11].
Definitions and general formulations
Let us consider the sorption of a single salt s from an aqueous solution by a membrane or ion exchange resin. A mole of the salt s is formed by ν1 ions 1 of electrical charge number z1 and ν2 ions of charge number z2, being νs (= ν1+ν2) the salt dissociation number and the electroneutrality condition ν1 z1 = - ν2 z2.
On the other hand, the membrane substance presents X equivalents of fixed electrical charges per unit of volume (X equiv./dm3) neutralised by 1/|z1| moles of counter-ions 1. Thus, the salt has the counter-ions 1 in common with the membrane and the co-ions 2 constitute the characteristic ions of the salt.
Moreover, the solvent water (index w) is present in both phases. The properties of the binary external solution are indicated with a bar ( ¯ ) over them. The external salt molality is ![]() , its molarity
, its molarity ![]() ,and the external water molarity is
,and the external water molarity is ![]() ; hence
; hence ![]() , where Mw is the molar mass of the solvent. Consequently, the external ionic concentrations are
, where Mw is the molar mass of the solvent. Consequently, the external ionic concentrations are
 | (1) |
On the other part, the membrane phase is a ternary one (membrane substance, salt and solvent) with an ion-exchange capacity X, a salt molarity cs and a water molarity cw. Here it is convenient to normalise the salt and water concentrations with respect to X:
 | (2) |
Finally, the ionic concentrations within the membrane are:
| (3) |
or, normalised with respect to X, they are:
| (4) |
The partition equilibrium is determined by the equality of the chemical potentials of the salt and of the solvent in both phases, i.e.
| (5) | |
| (6) |
At constant temperature and pressure, the degree of freedom is only one, the external salt concentration. Within the phase diagram, the system displaces along the solution/membrane coexistence curve in terms of ![]() , adjusting the membrane concentrations cs and cw ( or qw and qs) according to eqs.(5)-(6). In this work, we will only consider the equilibrium of eq.(5). Details of the water sorption determined by eq. (6) will be studied elsewhere.
, adjusting the membrane concentrations cs and cw ( or qw and qs) according to eqs.(5)-(6). In this work, we will only consider the equilibrium of eq.(5). Details of the water sorption determined by eq. (6) will be studied elsewhere.
Salt partition equilibrium
In order to analyse eq., the external salt potential ![]() is, as usually, given by:
is, as usually, given by:
| (7) |
where ![]() is the standard salt potential and
is the standard salt potential and![]() the mean ionic (stoichiometric) external molal salt activity coefficient. Similarly, the internal salt potential is expressed as
the mean ionic (stoichiometric) external molal salt activity coefficient. Similarly, the internal salt potential is expressed as
| (8) |
where ![]() is the standard potential and ys the corresponding molar salt activity coefficient in the membrane phase, being
is the standard potential and ys the corresponding molar salt activity coefficient in the membrane phase, being ![]() the membrane water content at . Both activity coefficients,
the membrane water content at . Both activity coefficients, ![]() and ys, are normalised at this limit
and ys, are normalised at this limit ![]() (cs → 0, qw→qow)
(cs → 0, qw→qow)
Equalising (7) and (8) one readily derives the expression of the partition equilibrium Ks. It results:
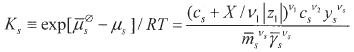 | (9) |
and, more practically,
 | (10) |
These thermodynamic expressions determine specific functionalities for the distribution of the salt between solution and membrane, i.e. ![]() or
or ![]() , with two unknown quantities. These are the partition constant Ks and the internal salt activity coefficient ys (if the external salt activity coefficient
, with two unknown quantities. These are the partition constant Ks and the internal salt activity coefficient ys (if the external salt activity coefficient ![]() is known).
is known).
These specific functionalities are determined by the characteristics of the salt/membrane system by (1) the ionic dissociation numbers (ν1, ν2) of the salt, and (2) which role is assumed by each ion, counter-ion and co-ion, being these roles established by the membrane. These specificities are readily put in evidence observing that from (10) the following limiting law at ![]() → 0 (qs « 1, ys → 1) is derived:
→ 0 (qs « 1, ys → 1) is derived:
| (11) |
This law clearly shows how the limiting partition of the salt is affected by the structure (ν1:ν2) of the salt and by the type of fixed charges (anionic or cationic) present in the membrane.
The experimental verification of (11) and, therefore, of the present approach, is easily achieved by plotting qs against the external molality ![]() and/or the external molal activity
and/or the external molal activity ![]() using log-log graphs. The theoretical exponents ns ( = 1+ν1/ν2) for different types of salts/membrane systems are the following:
using log-log graphs. The theoretical exponents ns ( = 1+ν1/ν2) for different types of salts/membrane systems are the following:
 | (12) |
Note that ν1:ν2 = |z2:z1|, where the second quotient is the usually used to typify electrolytes.
The law (11) can be analysed using experimental data taken from the literature. On one side, the systems MBr/MR (M = Na, Cs, Sr) studied by Meares et al. [12,13] are considered, being R is the membrane Zeo-Carb 315 (X = 0.548 mol/dm3 for NaR). In figure 1, qs is given in log-log plots versus ![]() (hollow symbols; concentration plots) and versus
(hollow symbols; concentration plots) and versus ![]() (filled symbols; activity plots). It is observed that the limiting law (11) (dashed line) is well obeyed, with ns = 2 for NaBr/NaR and CsBr/CsR and ns = 3/2 for SrBr2/SrR. These systems will be analysed with more details below, as the data sets are very abundant, covering an extended range of external salt concentration and allowing a complete study of eq.(10).
(filled symbols; activity plots). It is observed that the limiting law (11) (dashed line) is well obeyed, with ns = 2 for NaBr/NaR and CsBr/CsR and ns = 3/2 for SrBr2/SrR. These systems will be analysed with more details below, as the data sets are very abundant, covering an extended range of external salt concentration and allowing a complete study of eq.(10).

Fig. 1. NaBr//NaR, CsBr/CsR, SrBr2/SrR (Data, Meares et col. [12,13]
On the other hand, in figures 2 and 3 analogue graphs are given for systems of salts exhibiting other valences. As in figure 1, hollow symbols correspond to concentration plots and filled symbols to activity plots, being the number of points of these cases fewer, admitting only the analysis of the limiting law (11) .
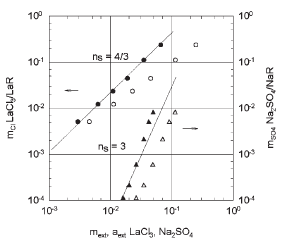
Fig. 2 . LaCl3/LaR, Na2SO4/NaR

Fig. 3. MgCl2/MgR, MgCl2/RCl
In figure 2 the molality of the co-ion within the membrane is plotted for (a) a 1:3 salt sorption system, LaCl3/LaR (R = Amberlite XE-100) [14], and (b) a 2:1 system, Na2SO4/NaR (R = Dowex 50W-X8, X = 4.60 meq/g) [15], in terms of external salt molality and activity. The slopes, ns = 4/3 and ns = 3, respectively, agree with the theoretical predicted values.
Finally, figure 3 presents the data, also the internal co-ion molality, of the sorption of the same salt MgCl2 by two different ion-exchange resins [16], representing examples for salts sorption systems of type 1:2 and 2:1, respectively. One is cationic (R = AG-50Wx2, X = 4.84 meq/g) and the other anionic (R' = AG-1x2, X = 4.60 meq/g). It should be noted that the roles of counter-ion and co-ion are interchanged from one example to the other. In both cases the values of the slopes, ns = 3/2 and ns = 3, respectively, correspond with the theoretical values.
For all cases the activity data applied are those given by Robinson-Stokes [17].
In all examples the limiting law is better verified in terms of the external salt activity, as it could be expected. Moreover, the original authors did not analyse their data, in any case, following the present approach.
Properties of the partition equilibrium
a) Determination of the thermodynamic partition constant
At the limit ![]() → 0 (qs → 0 or cs « X/ν1|z1|; ys → 1,
→ 0 (qs → 0 or cs « X/ν1|z1|; ys → 1, ![]() → 1) eq. (10) becomes
→ 1) eq. (10) becomes
 | (13) |
and herefrom it follows that it is convenient to define an apparent quantity kapp as
 | (14) |
Representing the first expression of kapp against the external salt molality ![]() (if
(if ![]() (
(![]() ) is unknown) in the concentration plots, as well as, the second expression (14) against the external salt activity
) is unknown) in the concentration plots, as well as, the second expression (14) against the external salt activity ![]() ( if
( if ![]() is known) in the activity plots, the extrapolation to
is known) in the activity plots, the extrapolation to![]() → 0 should give the same value, the value of the thermodynamic constant K´s.
→ 0 should give the same value, the value of the thermodynamic constant K´s.
In figure 4, these quotients are plotted for the systems of figure 1 in log-log graphs. It is observed that kapp has certainly a constant value at very low values of ![]() (hollow symbols) and of
(hollow symbols) and of ![]() (filled symbols), being this much more straightforward in the activity plot. The corresponding values of K´s are indicated in the graphs.
(filled symbols), being this much more straightforward in the activity plot. The corresponding values of K´s are indicated in the graphs.

Fig. 4. NaBr/NaR, CsBr/CsR, SrBr2/SrR (Data: Meares et col. [12,13].
b) Determination of the salt activity coefficient within the membrane phase
At higher salt concentrations, a deviation from the constant value is observed in the activity plot. Thermodynamically this fact indicates that the internal salt activity coefficient begins to be different from unity. Eq. (10) shows that, once the constant K´s is known, the activity coefficient ys of the salt within the membrane can be determined. Rearranging (10), it results:
 | (15) |
relation which enables to calculate ys for the three systems. The experimental values of these activity coefficients show a positive deviation (ys ≥ 1) in all three cases (see fig.5), being ys a function of the internal ionic composition of the membrane. As no theory is available for this dependency, several functions were tested. It is found that log (ys) varies linearly with the square root of the internal ionic salt strength, a guess certainly inspired in the properties of aqueous electrolyte solutions. This internal ionic salt variable, normalised with respect to X, the exchange capacity of the membrane, is defined as
| (16) |
being the empirical proposition for ys(Is):
| (17) |

Fig. 5. Log (internal salt activity coefficient) vs. Is 1/2
Figure 5 shows this linear relationship at lower Is. Moreover, it is interesting to point out that this dependency is coincident for both alkali ion (Na, Cs) systems. No theoretical foundation can be offered at the moment, but a direct application is at hand. Eqs. (10) and (13) include ys and hence log kapp should by linear in ![]() by eq. (17). Figure 6 illustrates these features using semi-log graphs. The linear extrapolation to Is → 0 gives the same results as in fig. 4.
by eq. (17). Figure 6 illustrates these features using semi-log graphs. The linear extrapolation to Is → 0 gives the same results as in fig. 4.

Fig. 6. Kapp vs. Is 1/2
Finally, it must be indicated that the peculiar properties of the internal salt activity coefficients noted in other cases (see Helfferich [7a]) are not observed in the present systems. Moreover, the different standard states used here and in the literature [7]-[8] should be taken into account.
General relation between the sorbed salt quantity qs and the external salt concentration ![]() (or
(or ![]() ).
).
Introducing the following operative quantity, k(![]() ),
),
 | (18) |
the expression (10) for the partition equilibrium can be rewritten as
| (19) |
Solving this equation for qs, the result is qs(![]() ), the general relationship between qs, the salt amount sorbed by the membrane, and the external salt molality
), the general relationship between qs, the salt amount sorbed by the membrane, and the external salt molality ![]() . Evidently, the functionality of this relation depends on the characteristic constitution factor (ν1:ν2) of each salt/membrane system.
. Evidently, the functionality of this relation depends on the characteristic constitution factor (ν1:ν2) of each salt/membrane system.
Hence, the cases considered in (12) must be analysed separately. The different equations corresponding to (19) are:
| (20) | |
| (21) | |
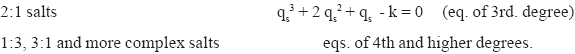 | (22) |
En general, the solutions of these equations for qs are functions of the independent term k and, by (18), of ![]() , or of
, or of ![]() , if the external activity coefficient
, if the external activity coefficient ![]() is not unitary. Even so, the calculus for qs is still direct as far as the internal activity coefficient ys has no effect. However, if this condition is to be relaxed (ys ≠ 1) at higher external concentrations, the solution for qs itself becomes more complex and must be attempted by successive iterations since k now includes a dependency on qs due to ys(qs) (see eqs.(16)-(17)). Nevertheless, if the later function is known (as by eq. (17)), the solution by iteration is always possible.
is not unitary. Even so, the calculus for qs is still direct as far as the internal activity coefficient ys has no effect. However, if this condition is to be relaxed (ys ≠ 1) at higher external concentrations, the solution for qs itself becomes more complex and must be attempted by successive iterations since k now includes a dependency on qs due to ys(qs) (see eqs.(16)-(17)). Nevertheless, if the later function is known (as by eq. (17)), the solution by iteration is always possible.
a) Sorption of symmetric salts (|z1| = |z2| ≡ z, νs =2)
This case is well known and has been extensively discussed in the literature (see p.ex. [7] and [8]), but in general considering only thermodynamically ideal systems. The corresponding equation (20) is a simple 2nd. degree equation and the general solution is:
 | (23) |
and
 | (24) |
where eqs. (9)-(10) have been taken into account.
In figure 1, the solid lines drawn through the data of the NaBr and CsBr systems have been calculated using these equations, taking into account the external as well as the internal salt activity coefficients, the latter within the linear range shown in figure 5. The experimental data are well reproduced. On the other hand, the dashed lines correspond to the ideal case without consideration of these activity coefficients, that is, to the limiting law
| (25) |
This law is readily obtained by a power expansion of the square root of (23) . It results:
| (26) |
Moreover, it is particularly interesting to state the expressions for the partition (distribution) coefficient pi (i = 1,2) among both phases for the ions themselves. These coefficients are readily derived from (24). To obtain more explicit expressions it is expedient to change the activity scale of the external solution from the molality scale to the molar scale by
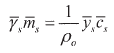 | (27) |
[17a] being ![]() the external molar activity coefficient of the salt and ρο the density of the pure solvent (water). Hence combining (24) and (27) it results:
the external molar activity coefficient of the salt and ρο the density of the pure solvent (water). Hence combining (24) and (27) it results:
 | (28) |
where k has been rewritten as
 | (29) |
Moreover, for very small concentration ![]() , the following limiting laws are obtained, which explicit the different characteristics of the partition of the two ions,
, the following limiting laws are obtained, which explicit the different characteristics of the partition of the two ions,
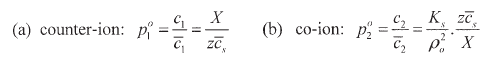 | (30) |
All these relations show that the relevant thermodynamic quantity for the salt sorption (electrolyte uptake) as well as for the ion exclusion is ![]() (sorption variable). The sorption of the salt is determined by the co-ion uptake (eqs.(28b)-(30b)) and, although this uptake increases with increasing external concentration (normal sorption effect), the fixed ion concentration X has the opposite effect. As the co-ion and the fixed ion of the membrane have the same electrical charge sign, with increasing X (higher ion-exchange capacity of the membrane) the salt sorption decreases, that is, the external electrolyte is increasingly excluded form the membrane (ion exclusion effect) with higher X. By the same effect the characteristic sorption quantity k (eq. (29)) decreases with increasing X. On the other hand, the marked asymmetry of the counter-ion distribution increases with increasing X (eqs.(28a)-(30a)), but decreases with increasing external concentrations.
(sorption variable). The sorption of the salt is determined by the co-ion uptake (eqs.(28b)-(30b)) and, although this uptake increases with increasing external concentration (normal sorption effect), the fixed ion concentration X has the opposite effect. As the co-ion and the fixed ion of the membrane have the same electrical charge sign, with increasing X (higher ion-exchange capacity of the membrane) the salt sorption decreases, that is, the external electrolyte is increasingly excluded form the membrane (ion exclusion effect) with higher X. By the same effect the characteristic sorption quantity k (eq. (29)) decreases with increasing X. On the other hand, the marked asymmetry of the counter-ion distribution increases with increasing X (eqs.(28a)-(30a)), but decreases with increasing external concentrations.
Summarising, in the present case of symmetric salts the sorption of the external salt is proportional to the square of the external concentration and inversely proportional to the square of the fixed charge concentration, while the partition of the salt is not constant, but directly proportional to the external concentration and inversely proportional to X. Finally, with higher valence z the salt sorption increases and the ion exclusion is less efficient.
b) Sorption of 1:2 and 2:1 ternary salts ![]()
This case has not been discussed in the literature, so far the author is aware. As indicated by eqs.(21)-(22) the sorption of both types of ternary salts is governed by relations of 3rd degree, notwithstanding that they present very different sorption characteristics, as it is stated by eq. (12) and as it has been corroborated by the experimental examples given in Section 4.
The solutions of the 3rd degree relations depend on the values of the coefficients of the powers of qs and if the corresponding discriminant Q of the cubic form is positive or negative [19]. Let a general cubic form be ![]() , then the cubic discriminant Q is given by
, then the cubic discriminant Q is given by ![]() , where the quantities p and q are defined by
, where the quantities p and q are defined by ![]() and
and ![]() . If (a) Q>0 the solution of the cubic equation is given by the Cardano- Tartaglia (CT) formula, and if (b) Q < 0 the solution is a trigonometric one (casus irreducibilis, CI) [19]. Both cases apply to the sorption systems at hand. As it is readily verified, the sorption of 2:1 salts corresponds to case (a), while case (b) is pertinent for the sorption of 1:2 salts. Both cases will be discussed separately below.
. If (a) Q>0 the solution of the cubic equation is given by the Cardano- Tartaglia (CT) formula, and if (b) Q < 0 the solution is a trigonometric one (casus irreducibilis, CI) [19]. Both cases apply to the sorption systems at hand. As it is readily verified, the sorption of 2:1 salts corresponds to case (a), while case (b) is pertinent for the sorption of 1:2 salts. Both cases will be discussed separately below.
2:1 salts ![]() : The sorption of these salts follow the cubic eq. (22), i.e.
: The sorption of these salts follow the cubic eq. (22), i.e.
| (31) |
Hence p = - 1/3 and q = - 2/27 - k and the discriminant Q is positive:
 | (32) |
Accordingly the solution of (22) is the Cardano-Tartaglia formula (CT) [19], i.e.
| (33) |
In the present case it takes the following explicit form:
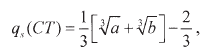 | (34) |
where
 | (35) |
Examples of this sorption case are the systems Na2SO4/NaR (cationic resin) and MgCl2/ ClR´ (anionic resin) discussed in Section 4, as representatives of the simplest systems of co-ions of higher valence than that of the counter-ions. These systems are analysed in Figures 2 and 3 and it is verified that the limiting law, is cubic, i.e.
| (36) |
as it is indicated by (11)-(12). This relation follows also directly from (31), but it is not inferred directly from the general formula (34). Moreover, the application of this formula is cumbersome and a general power series expansion in terms of k, as in (26), is also adequate here. Analytically such expansion is quite difficult, but is can be obtained with the aid of a computer program [20]. The result is the following:
| (37) |
expression which confirms the limit (36).
Numerically (37) is not very satisfactorily, as with the nine terms shown it just reproduce qs at k=0.15 within 2%. On the other hand, considering only the two first terms, i.e.
| (38) |
the expansion is valid up to k = 0.01 within 0.1%. Hence it seems practical to look for a least squares expansion of four terms with the first two terms fixed. However, for the range ![]() , this yields an expression (1) which, within the last decade (
, this yields an expression (1) which, within the last decade ( ![]() ), has a very poor reproducibility (an appreciable wriggling). Therefore, looking for another simple functionality it is found that a rational expression such as
), has a very poor reproducibility (an appreciable wriggling). Therefore, looking for another simple functionality it is found that a rational expression such as
 | (39) |
with two constants adjusted by least squares, proves to be more suitable and precise in the same range (![]() ). Eq. (39) replaces satisfactorily the general solution (34)-(35) up to k = 1 and at low k-values it becomes the limiting series (38).
). Eq. (39) replaces satisfactorily the general solution (34)-(35) up to k = 1 and at low k-values it becomes the limiting series (38).
The corresponding ion partition expressions for this case are the following:
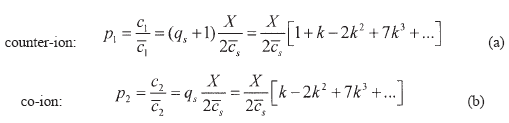 | (40) |
with 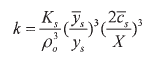 | (41) |
and the limiting relations are
 | (42) |
Thus, according to the foregoing results, in this case of co-ions of higher valence than that of the counter-ions the electrolyte uptake is, through the sorption quantity k (eqs. (31) and (41)), proportional to the 3rd power of the sorption variable ![]() . That is, the salt sorption is here proportional to the 3rd power of the external concentration, but it is opposed by the 3rd power of the fixed charge concentration. Additionally the partition of the salt is again not constant, but is directly proportional to the square of
. That is, the salt sorption is here proportional to the 3rd power of the external concentration, but it is opposed by the 3rd power of the fixed charge concentration. Additionally the partition of the salt is again not constant, but is directly proportional to the square of ![]() and inversely proportional to the square of X. Consequently, at low external concentrations with co-ions of higher valence the salt sorption decreases and the ion exclusion is more efficient.
and inversely proportional to the square of X. Consequently, at low external concentrations with co-ions of higher valence the salt sorption decreases and the ion exclusion is more efficient.
Further comments will be made in combination with the results of the next case at the end of this section.
1:2 salts : ![]() The salt uptake follows now the cubic eq. (21), i.e.
The salt uptake follows now the cubic eq. (21), i.e.
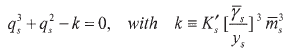 | (43) |
where the linear term is missing. Since here p'= -1/3 and q'= 2/27 -k, the discriminant Q´ is negative if k < 4/27:
 | (44) |
Accordingly the solution (33) takes in this case a complex form (casus irreducibilis, CI):
 | (45) |
where
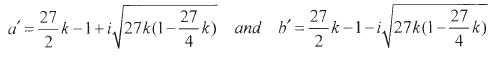 | (46) |
but, as the imaginary terms cancel each other, the solution is real. Analytically, in a trigonometric formulation (2), it can be shown[19] that eq. (45) equivalent to
| (47) |
Numerically both expressions give the same real results over the whole range of k. Moreover, it is interesting to state that qs = 1/3 at Q'=0 (k =4/27 » 0.15).
This solution applies to the systems SrBr2/SrR of figure 1 and MgCl2/MgrR of figure 3, systems which are representatives of the simplest case of counter-ions of higher valence than that of the co-ions. The solid lines of this case in figure 1 have been calculated using (47) taking into account both activity coefficients, the internal one within the linear range of figure 5; the experimental data are well reproduced. Furthermore, for these systems the dashed line in figure 1 and the solid line in figure 3, respectively, represent the ideal calculus with unitary activity coefficients according to the following limiting law
| (48) |
law which is directly obtained from (43), but which cannot be derived directly from the general solutions (45)-(47).
As in the preceding case, the application of the general solutions (45)-(47) is complicated and a power series expansion in k is also recommendable. However, analytically such expansion is in this case even more involved because of the peculiar behaviour of the function arc cos at (-1), but the computer program [20] again solves the problem. The series is the following:
| (49) |
expansion which confirms the limits (48).
Numerically (49) is, as (37), not very satisfactorily, as, using the nine terms given, it only reproduce qs at k=0.1 within 2%. On the other hand, with only the two first terms, i.e.
 | (50) |
the expansion is valid up to k=0.01 within 0.5% and a least squares representation of four terms with these first two terms fixed yields here a good formula within 0.5% for the range ![]() :
:
 | (51) |
Furthermore, looking also for a rational expression for the same range ![]() it is found that the simple expression with only one adjustable parameter
it is found that the simple expression with only one adjustable parameter
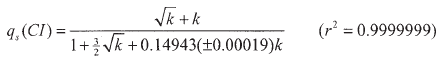 | (52) |
is again much more effective, with an error of only 0.1%. and is a very good substitute for the solution (47). Also, this relation is compatible at low k with the limiting expression (50).
Finally the ion partition expressions are in this case:
|
| (53) |
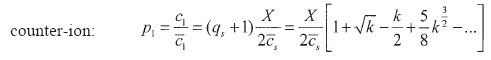

being these expressions reduced to the following limiting laws for ![]() = X / 2
= X / 2
 | (54) |
where eq. (27) (or (41)) has again been taken into account.
Thus, according to all these relations, in this case of counter-ions of higher valence than that of the co-ions, which is the inverse of the preceding case, the salt uptake behaviour is completely different. It is now proportional to the square root of the sorption quantity k (eqs.(48)- (49)), i.e. it is only proportional to the power 3/2 of the sorption variable ![]() . Hence the electrolyte sorption follows now a much less pronounced dependence (with power 3/ 2) on the external concentration, and it is also much less opposed (also with power 3/2) by the exchange capacity of the membrane. Moreover, the partition of the salt is again not constant, but much less concentration dependent; it is now directly proportional to the square root of
. Hence the electrolyte sorption follows now a much less pronounced dependence (with power 3/ 2) on the external concentration, and it is also much less opposed (also with power 3/2) by the exchange capacity of the membrane. Moreover, the partition of the salt is again not constant, but much less concentration dependent; it is now directly proportional to the square root of ![]() and inversely proportional to the square root of X. Therefore, at low external concentrations with counter-ions of higher valence the salt sorption is higher and the ion exclusion is less efficient than in the two preceding cases.
and inversely proportional to the square root of X. Therefore, at low external concentrations with counter-ions of higher valence the salt sorption is higher and the ion exclusion is less efficient than in the two preceding cases.
Finally, some general comments are to be made. On one hand, it has been shown that the appropriate sorption variable is in all cases ![]() . The general features of this variable and of the characteristic sorption quantity k stated for the symmetric salts are quite general: the salt sorption is always directly proportional to the external concentration and inversely proportional (ion exclusion) to the internal fixed charge concentration. But these dependencies change with the stoichiometry (ion valences) of the sorption system (see below), i.e. the change of the valence of one of the ions determines a completely different behaviour of the sorption system (which is essentially due to the change of the exponent in (11)-(12)). So the sorption-system characterising structural quotient ν1 / ν2 determines the different sorption behaviours given by eqs.(23), (34) and (47). Furthermore, the co-ion relations (28b), (40b) and (53b) describe quantitatively the partition (and sorption) of the salt by the membrane, quantities which are always external concentration dependent. Finally, the quite different limiting laws (25), (36) and (48) as well as (30b), (42b) and (54b) are in complete accordance with the general expression (11).
. The general features of this variable and of the characteristic sorption quantity k stated for the symmetric salts are quite general: the salt sorption is always directly proportional to the external concentration and inversely proportional (ion exclusion) to the internal fixed charge concentration. But these dependencies change with the stoichiometry (ion valences) of the sorption system (see below), i.e. the change of the valence of one of the ions determines a completely different behaviour of the sorption system (which is essentially due to the change of the exponent in (11)-(12)). So the sorption-system characterising structural quotient ν1 / ν2 determines the different sorption behaviours given by eqs.(23), (34) and (47). Furthermore, the co-ion relations (28b), (40b) and (53b) describe quantitatively the partition (and sorption) of the salt by the membrane, quantities which are always external concentration dependent. Finally, the quite different limiting laws (25), (36) and (48) as well as (30b), (42b) and (54b) are in complete accordance with the general expression (11).
On the other hand, the counter-ion exhibits common aspects. It follows at low concentrations, in all cases, the same high and asymmetric distribution relation (eqs.(30a), (42a) and (54a), which is directly given by the inverse of the sorption variable. This assymmetry increases with increasing X, but decreases with increasing ![]() , presenting only at higher
, presenting only at higher ![]() different behaviours (eqs.(28a), (40a) and (53a).
different behaviours (eqs.(28a), (40a) and (53a).
Concluding remarks
In his book [7b] Helfferich stated qualitative ´general rules´ for the electrolyte uptake by ion-exchange systems which are confirmed and complemented by the present formulation. However, Helfferich´s conclusions are derived using concepts such as individual ionic chemical potential and single electrical (Donnan) potential differences and a swelling pressure (difference) for the description of the equilibria at the solution/membrane interface, quantities which are all thermodynamically undefined (unmeasurable). Their use is an old controversial issue; they finally cancel or must be combined to give measurable quantities corresponding to independent components in the sense of Gibbs´ phase rule.
Instead, our formulation obviates these electrical and pressure aspects working directly in terms of variables of the phase rule, recognising the ions only as constituent parts of the salts (eqs.(7)-(8)). The resulting thermodynamic relations indicate directly the characteristic features of these phase equilibria, properties which are verified experimentally. Notwithstanding all this, our results and conclusions coincide with those of Helfferich. Strictly speaking, the problem at hand is only a particular, although very important, case of a phase equilibria involving electrolytic components with common ions with one ionic constituent (the membrane matrix) restricted to be present in only one of two phases involved.
To state the general rules it is convenient to compare the two last cases studied with the case of symmetric salts, in particular with uni-univalent electrolytes (z=1). Then, for a same type of membrane, the 2:1 case corresponds, as has been stated, to a salt with a co-ion of higher valence, while the 1:2 case corresponds to a salt with a counter-ion of higher valence. So, given the cationic membrane system NaBr/NaR (1:1) appropriate ternary systems are, for example, SrBr2/SrR (1:2) and Na2SO4/NaR (2:1), respectively, systems which have been characterised in the previous sections.
Considering the pertinent sorption equations (23), (34) and (47) one finds immediately that, other things being equal, the following sorption sequence:
 | (55) |
i.e. the salt uptake is increased by counter-ions of high valence and by co-ions of low valence (the sorption of SrBr2 is higher than that of NaBr and that of this salt is higher than that of Na2SO4). On the contrary, ion exclusion is more efficient with counter-ions of low valence and co-ions of high valence (Na2SO4 is more strongly excluded than NaBr and this salt more strongly than SrBr2).
In general the salt sorption (and partition) is, as it should be, a direct function of the external salt concentration, but this solubility is inhibited by an increasing exchange capacity of the membrane. Accordingly the partition is always very low at low concentrations and rises with increasing external concentration and, thereby, the salt sorption isotherms have always a strong positive curvature. Or, the other way round, the efficiency of electrolyte exclusion increases with decreasing external concentration and with increasing exchange capacity.
In conclusion, the direct thermodynamic formulation allows to describe satisfactorily these sorption systems, in particular those of non-symmetric electrolytes. In the case of symmetric salts our results coincide and complement those of previous approaches.
Finally, another general aspect is to be pointed out. All the ion partition coefficient expressions given above show a dependency on the external salt concentration; that's to say that these coefficients are not constant and vary with ![]() . On the other hand, in Biophysics [5] the very popular membrane potential approach due to Goldman-Hodgkin-Katz (GHK) [3]-[4] assumes that they are constant. The present paper shows that this premise is incorrect and this feature is one of the reasons why the GHK potential formula do not reproduce correctly the experimental membrane potential data [18]. Moreover, the GHK formulation fails for non-symmetric electrolytes, while the thermodynamic approach works quite well [18].
. On the other hand, in Biophysics [5] the very popular membrane potential approach due to Goldman-Hodgkin-Katz (GHK) [3]-[4] assumes that they are constant. The present paper shows that this premise is incorrect and this feature is one of the reasons why the GHK potential formula do not reproduce correctly the experimental membrane potential data [18]. Moreover, the GHK formulation fails for non-symmetric electrolytes, while the thermodynamic approach works quite well [18].
Acknowledgement
The assistance of Cristian Muzzio for the use of the Mathematica program is gratefully acknowledged.
The author is most indebted to Prof. H. J. Schumacher for all his support, in particular for giving a direct and definitive orientation to his academic career
Notas
(1) ![]()
(2) ![]()
References
[1] Schloegl,R., Stofftransport durch Membranen, Dr. D. Steinkopff Verlag, 1964. [ Links ]
[2] Teorell, T., Progress in Biophysics 1953, 3, 305. [ Links ]
[3] Goldman, D., J.Gen.Physiol. 1943, 27, 37. [ Links ]
[4] Hodgkin, A.; Katz, B., J.Physiol. 1949, 108, 37. [ Links ]
[5] Hille, B., Ionic channels of excitable membranes, 2nd ed., Sinauer Assoc., 1991. [ Links ]
[6] Nobel, P., Physicochemical and Environmental Plant Physiology, Academic Press, 1991. [ Links ]
[7] Helfferich, F., Ion exchange, McGraw-Hill, 1962; (a) p.143, (b) sect. 5-3c. [ Links ]
[8] Laskminarayanaiah, L., Transport phenomena in membranes, Academic Press, 1969. [ Links ]
[9] Donnan, F.G., Z.Elektrochemie 1911, 17, 572; [ Links ] Donnan, F.G.; Guggenheim, E.A., Z.Phys. Chemie 1932, 162, 346. [ Links ]
[10] Tanford, C., Physical Chemistry of macromolecules, Wiley, 1961. [ Links ]
[11] Timmermann, E.O., Transport phenomena through ionic membranes due to pressure, concentration and electrical potential differences: theoretical and experimental interpretation using phenomenological conductance coefficients, III Congreso Ibero- Americano en Ciencia y Tecnología de Membranas (CITEM III), September 14-15, 2001, Aveiro, Portugal. [ Links ]
[12] Mackie, J.S.; Meares, P., Proc. Roy. Soc. 1955, A232, 485. [ Links ]
[13] Meares, P.; Thain, J.F., J.Phys.Chem. 1968, 72, 2789. [ Links ]
[14] Gustafson, R., J. Phys. Chem. 1966, 70, 957. [ Links ]
[15] Boyd, G.E.; Bunzl, K., J. Am. Chem. Soc. 1967, 89, 1776. [ Links ]
[16] Freeman, D.H.; Patel, V.C.; Buchanan, T.M., J.Phys.Chem. 1965, 69, 1477. [ Links ]
[17] Robinson, R.A.; Stokes, R.H., Electrolyte solutions, 2nd edition, Butterworths, 1959, (a) p.32. [ Links ]
[18] Timmermann, E.O., Membrane potential: experimental analysis of its thermodynamic expression and of other formulations, II Congreso Ibero-Americano en Ciencia y Tecnología de Membranas (CITEM II), August 8-10, 1994, Rio de Janeiro, Brasil. [ Links ]
[19] (a) Gellert, W.; Kuenstner, H.; Hellwichu, M.;. Kaestner, H. (eds.), Grosses Handbuch der Mathematik, Buch u. Zeit Verlagsges. GmbH, 1969; [ Links ] (b) Sagastume Berra, A.E.; Fernández, G., Algebra y cálculo númerico, ed. Kapelusz, 1960. [ Links ]
[20] Mathematica 4 - version 4.0.0.0 - Wolfram Research, Inc. [ Links ]

