










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Asociación Química Argentina
versión impresa ISSN 0365-0375
An. Asoc. Quím. Argent. v.93 n.4-6 Buenos Aires ene./dic. 2005
REGULAR PAPERS
Self-assembly of platinum nanowires on HOPG
Quaino, P.M.1; Gennero de Chialvo, M.R.1; Vela, M.E.2; Salvarezza, R.C2*
1PRELINE, FIQ, Universidad Nacional del Litoral, Sgo. del Estero 2829, S3000AOM, Santa Fe, Argentina
2INIFTA, CONICET, Universidad Nacional de La Plata, Suc. 4, C.C. 16, B1906ZAA La Plata, Argentina. Fax: +54-221-4642, E-mail: robsalva@inifta.unlp.edu.ar
Received December 8th, 2005. In final form December 20th, 2005
Dedicated to the memory of the late Prof. Hans J. Schumacher on the occasion of his 100th birthday
Abstract
Platinum nanoparticles were obtained on highly oriented pyrolytic graphite (HOPG) in 0.01 M H2Cl6Pt and 0.1 M HCl solution by open circuit deposition. The resulting structures were characterized electrochemically by cyclic voltammetry and morphologically by ex situ scanning tunneling microscopy. The deposition on the substrate occurs preferentially at step edges instead of flat terraces. The density of particles as well as their dimension depend on the time of immersion in the hexachloroplatinic solution. Pt nanowires are formed for a 3 h immersion time whereas for a 90 s immersion the typical clusters are mainly hemispherical.
Resumen
Se obtuvieron nanocintas de platino sobre bordes de escalón de grafito pirolítico altamente orientado (HOPG). La deposición se llevó a cabo a circuito abierto en una solución 0.01 M de H2PtCl6 y 0.1 M de HCl. Las estructuras resultantes fueron caracterizadas electroquímicamente mediante voltametría cíclica y morfológicamente mediante microscopía de efecto túnel ex-situ. Tanto la densidad de partículas y como su dimensión dependen del tiempo de inmersión en la solución del complejo hexacloroplatínico.
Introduction
Nanoparticles are particularly interesting in research due to their different physicochemical properties with respect to the bulk materials. They have better properties for many applications in fields such as catalysis [1], electronics [2], and biodiagnostics [3].
From the point of view of surface phenomena, and in particular those processes involving adsorbed species, it is evident that when the particle size diminishes (< 10 nm), the number of surface atoms with low coordination number increases. Accordingly the catalytic activity of nanoparticles is considerably different to that of the bulk material.
Nanoparticles and clusters supported on many materials (oxides, zeolites, etc) have been studied in the field of heterogeneous catalysis [4]. In particular, in the field of electrocatalysis, metal nanoparticles dispersed on conductive substrates have been used as electrodes for different applications. In this sense, carbon-supported platinum nanoparticles constitute a typical system due to their technological significance in the fabrication of electrodes for PEM fuel cells [5].
Electrodeposition is one of the methods used for the preparation of Pt nanoparticles on carbon substrates. For this reason the study of Pt electrodeposition on different types of carbon electrodes has attracted considerable attention. Zubimendi et al. [6] studied the first stages of Pt electrodeposition on HOPG (highly oriented pyrolytic graphite) using ex-situ STM. They observed the 3D nucleation and growth of Pt on surface defects, in particular on step edges. This 3D growth follows the Volmer-Weber mechanism. Pt clusters of 2 nm were observed at low cathodic potentials and low electrodeposition charge density. These clusters have a tendency to form rows of nucleus aligned in well defined directions forming ordered 3D structures. Compact Pt crystals are formed at more cathodic potential and higher electrodeposition charge densities. Similar results have been reported by Lee et al. [7]. They also found that 3D growth starts before a Pt monolayer is formed, following the Volmer-Weber mechanism. Particles of 10-20 nm in diameter are formed first and surface diffusion occurs later with neighboring particles stuck together as deposition time increases. These particles have the tendency to form aligned rows in a 2D order.
Electrochemical preparation of Pt nanoparticles on HOPG with size selectivity is explained by Zoval et al. [8]. They describe a method to obtain Pt nanocrystals dispersed on HOPG from hexachloroplatinic acid using a potentiostatic pulse at high overpotentials. Particles obtained by this procedure are highly monodisperse (for diameters less than 5.0 nm), stable in air during many days and, on average, well spaced on the HOPG surface. They also prepared Pt nanocrystals by electroless deposition. In this case they are ring-shaped and not uniformly distributed on the graphite basal plane surface.
There are a special type of nanoparticles characterized by having only two nanoscopic dimensions. They are called nanowires when their diameters are less than 100 nm, being able to reach a length of several microns. This characteristic gives particular properties to the nanowire, as the variation of its conductivity induced by the surface adsorption of molecules, e.g. thiols on Cu and Au [9]. Platinum nanowires on HOPG have been prepared by several methods such as chemical vapor deposition [10], self assembly of Chini clusters [11], cyclic voltammetry deposition [12] and potential deposition programs [13]. Metal mesowires (diameters greater than 100 nm) were also obtained on HOPG to be used as elements for building different types of sensors, such as Pd mesowires used as hydrogen sensors [14]. In this case step edges act as templates for the growth of the metal mesowires.
In the present work, the formation of platinum nanowires on HOPG by open circuit deposition is described and discussed. The resulting structures are characterized electrochemically by cyclic voltammetry and morphologically by ex situ scanning tunneling microscopy.
Experimental
Preparation of Pt nanowires
Platinum nanoparticles were formed on a HOPG substrate by spontaneous deposition. The substrate consisted in a HOPG SPI foil, which were mounted on a special device in order to be used as a rotating electrode. It was immersed during different times in a 0.01 M hexachloroplatinic acid solution prepared from dissolution of the solid H2Cl6Pt in 0.1 M HCl solution. Since no external potential is applied to the interface, the deposition of Pt particles takes place at open circuit potential (OCP), which was measured against a hydrogen reference electrode in a 0.1 M de HCl solution free of Pt ions.
The immersion time was ranged between 10 s to 3 h in order to vary the surface density of particles. Once prepared the Pt-HOPG electrode was removed from the deposition solution and rinsed with thrice distilled water.
STM measurements
Ex situ characterization of the Pt particles on HOPG was made by a Nanoscope III (Digital Instruments) Scanning Tunneling Microscope (STM) using commercial Pt-Ir nanotips. Typical conditions of imaging were 600 < Ebias / mV < 800 and 120 < itun / pA < 250. These values ensure a sample-tip distance that prevents dragging of the particles.
Electrochemical measurements
Cell description: Electrochemical measurements were made in a three electrode Pirex glass cell specially designed to be used with a rotating disc electrode. Figure 1 a and b show an scheme of the cell and the rotating electrode holder, which allowed to obtain a reproducible working electrode area and to eliminate problems due to the degassing procedure.

Figure 1: a) Electrochemical cell: (1) rotating disk electrode holder, (2) gas inlet, (3) auxiliary electrode, (4) and (5) bubbling tubes, (6) Luggin capillary (7) reference electrode, (8) reference electrode holder. b) Rotating electrode holder system (1) connection to the motor, (2) HOPG electrode, (3) Teflon body.
The working electrode was a HOPG substrate with the Pt nanoparticles deposited as described above. The counterelectrode was a large area Pt wire and the reference electrode consisted of a hydrogen bubble immersed in the same electrolyte solution. Therefore, all potentials in this work are referred to the reversible hydrogen electrode (RHE).
Voltammetric characterization: The electrochemical characterization of the Pt-HOPG electrode was carried out using cyclic voltammetry. Runs were made in a 0.5 M H2SO4 electrolytic solution, at a sweep rate of 0.1 V s-1 under nitrogen gas bubbling to avoid the interference of dissolved oxygen and at a temperature of 30 ºC.
Voltammetric profiles were obtained for both the HOPG substrate and the Pt-HOPG electrode in the range -0.5 ≤ E / V ≤ 1.2. A potenciostat-galvanostat Radiometer PGP201 controlled by an acquisition-generation data card Advantech PCL-818 and the software Labtech Notebook© were used. All voltammograms were recorded at a rotation rate of 8100 rpm, which was controlled by a Tacussel EDI 10000 rotating disk equipment.
Results
Electrochemical studies
The spontaneous deposition of platinum particles on HOPG was followed through the dependence of the open circuit potential (EOCP) on time. It can be observed in Figure 2 one of these responses, where the value EOCP = 0.84 V was obtained. All the experiments carried out indicate that the open circuit potential was always comprised between 0.8 ≤ EOCP / V ≤ 0.9, being approximately constant with time.

Figure 2: Open circuit potential vs. time during the immersion of HOPG in 0.01 M H2Cl6Pt and 0.1 M HCl 0.01 M solution.
The voltammetric response of the HOPG blank electrode in 0.5 M H2SO4 in the range - 0.05 < E / V < 1.2 can be observed in Figure 3a, exhibiting the typical response of a capacitive interfacial process. Note that no hydrogen evolution (her) is detected at the cathodic limit. On the other hand, the presence of deposited Pt particles on the HOPG surface modifies the electrochemical profile. In fact, the voltammetric curve of the Pt modified HOPG electrodes exhibits an increasing cathodic current starting at 0.0 V (Figure 3b), which indicates that the small amount of Pt on the HOPG surface is able to catalyze the HER reaction. When the immersion time is increased to 90 s (Figure 4), the voltammogram exhibits an enhancement of the hydrogen evolution current as well as the appearance of an incipient hydrogen adsorption region. This behavior can be observed more clearly in the potentiodynamic response corresponding to an immersion time of 3 h (Figure 5), where the higher density of Pt particles produces a voltammetric profile similar to that of a polycrystalline platinum electrode.

Figure 3: Cyclic voltammograms in 0.5 M H2SO4 at 0.025 V s-1. (a) HOPG electrode, (b) Pt-HOPG electrode. Immersion time: 10 s.

Figure 4: Cyclic voltammogram of a Pt-HOPG electrode in 0.5 M H2SO4 at 0.025 V s-1. Immersion time: 90 s.
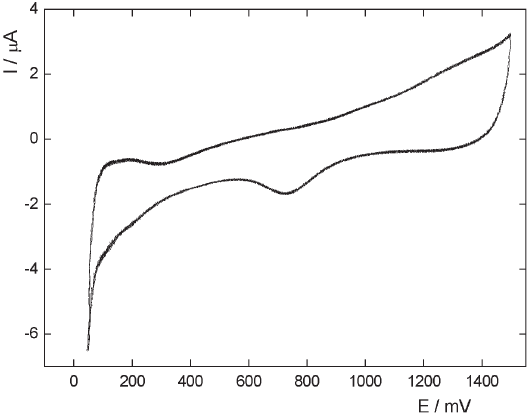
Figure 5: Cyclic voltammogram of a Pt-HOPG electrode in 0.5 M H2SO4 at 0.025 V s-1. Immersion time: 3h.
STM studies
Typical STM images of the HOPG substrate are shown in Figure 6 (a and b). Flat terraces of 1 - 2 µm wide are observed separated by 0.35 nm steps in height that correspond to the interlayer spacing of the HOPG. Higher resolution of the STM images (Figure 6b) reveals the hexagonal lattice with nearest neighbor distance d = 0.242 nm where three of the six C atoms of the well-known surface structure of graphite is imaged.


Figure 6: STM images taken at Ebias= 30 mV and Itunn= 15 nA on HOPG. a) 2.56x2.56 µm. b) 5x5 nm.
The HOPG substrate after 3 h immersion in the hexachloroplatinic acid solution exhibits preferential deposition of Pt particles along step edges (Figure 7a). Nucleation of Pt adatoms at step edges is favored by a higher coordination number at these sites. The growth along the steps forms Pt "wires" of 7 nm in width (25 atoms) and 0.7 nm in height (≈3 atomic layers), as it can be observed in Figure 7b-c. The wire is formed by quasi hemispherical small Pt nanoparticles ≈2- 3.5 nm in size. Other row of disconnected nanoparticles running parallel to the nanowire is also observed. Although most of the nanoparticles grow at steps some of them also nucleate on terraces, probably on surface defects.

Figure 7: STM images of Pt deposited on HOPG in 0.01 M H2Cl6Pt and 0.1 M HCl 0.01 M solution. Immersion time: 3 h. Ebias= 729 mV and Itunn= 123 pA. a) 263x263 nm. b) 20x20 nm.
Decreasing the immersion time to 90 s produces a small amount of Pt deposited at the active sites of the HOPG surface (Figure 8a-c). In this case the typical shape and size of the particles is more regular. They are mainly hemispherical with a diameter of 3 nm and 0.3-0.5 nm in height (≈1-2 monolayers). Three rows of Pt particles are observed and the third still "sees" the direction of the HOPG step but is 0.38 nm apart from the second. The STM images corresponding to the deposition carried out at an immersion time of 10 s are similar to those described for 90 s, with a less amount of particles.

Figure 8: STM images of Pt deposited on HOPG in 0.01 M H2Cl6Pt and 0.1 M HCl 0.01 M solution. Immersion time: 90 s. Ebias= 1.01 V and Itunn= 204 pA. a) 250x250 nm. b) 32x32 nm.
Discussion
The present work has demonstrated experimentally that when a freshly exfoliated HOPG flake is immersed in a 0.01 M H2Cl6Pt and 0.1 M HCl solution, the deposition of platinum nanowires can be carried out in absence of an external flow of electric charge. The deposition process is verified in the step edges originated during the HOPG exfoliation and the corresponding open circuit potential remains approximately constant during the immersion. The values of this potential varied in the range 0.8 ≤ EOCP / V ≤ 0.9 for the different experiments carried out, probably due to the different degree of oxidation of functional groups present in the surface defects of the substrate. Furthermore as the EOCP values are very high, it can be concluded that the overpotential involved in the platinum reduction reaction should be quite low. This fact favors the deposition on the steps and allows the development of nanowires before the deposition on
the terraces can take place.
It should be noticed that, conversely to the results corresponding to the application of electrodeposition processes [8, 15], in this case there is a high specificity in the platinum nucleation sites. On the contrary, the results of Zoval et al. obtained in similar experimental conditions show that the deposition process is verified on the whole surface of the HOPG substrate, although they used a more diluted platinum solution (0.001 M H2Cl6Pt and 0.1 M HCl) and the open circuit potential is not indicated [8]. Similar results were obtained by Rodriguez-Nieto et al. by electrodeposition in 0.5 M H2SO4 solution [15].
Ideally, the HOPG substrate cleaved in air provides clean large flat terraces containing carbon atoms in the hexagonal lattice array, with no other functional groups present, with the exception of the step edges. Nevertheless, the STM observations show that the terraces present a large amount of defects, which are partially oxidized. When such surface is immersed in a solution containing Cl6Pt2- anions with an excess of chloride ions the step edges act as an electron source, which can produce through a sequence of elementary steps the complete reduction of the hexachloroplatinate anions,
| PtCl62- + 4e → Pt + 6Cl | (1) |
This reaction is favored by the open circuit potential, which is highly anodic with respect to the zero charge potential (≅ 0.0 V [16]) and is produced almost exclusively at the step edges. Consequently, the origin of the electrons needed in equation (1) must be justified, as well as the preferential deposition in the steps. Related to the first item Zoval et al. pointed out that the electrons are supplied by incompletely oxidized functionalities (aldehydes, alcohols, etc.) existing at defects on the HOPG surface [8]. Nevertheless, the amount of such functional groups would not be enough to obtain nanowire structures such as those illustrated in Figures 7-8. Consequently, another source of electrons should be considered. In this sense, the explanation given by Simonov et al. sounds quite reasonable. They stated that the reduction of metal ions involves free electrons of the carbon matrix [17]. Therefore, in a solution with excess of chloride ions as that used in the present work, there is an excess of negative charge in the double layer which produces the displacement of the free electrons from the surface terraces towards the step edges allowing the occurrence of reaction (1).
Finally, it is known that the specific rate constant for the step reaction is significantly greater than that corresponding to the reaction taking place on the terraces [18]. Therefore, as the nucleation process is initiated on the step edges, the development of the nanowire structure in this localized place is favored.
Conclusions
Pt nanoparticles were deposited on an HOPG substrate at open circuit potential in a 0.01 M H2Cl6Pt and 0.1 M HCl solution. The deposition on the substrate occurs preferentially at step edges instead of flat terraces. The density of particles as well as their dimension depend on the time of immersion in the hexachloroplatinic solution.
Pt "wires" of 7 nm in width (25 atoms) and 0.7 nm in height (approximately 2 atomic layers) are formed for a 3 h of immersion time whereas for a 90 s of immersion the typical clusters have a diameter of 3 nm and a height of 0.3-0.6 nm. The voltammetric response in the cathodic potential hydrogen region is in accordance with the area of the Pt clusters observed by STM.
Acknowledgements
Financial support from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) PICT 2003-06-17492 and the Universidad Nacional del Litoral is gratefully acknowledged. MEV is a member of the research career of CIC, Province of Buenos Aires.
References
[1] Li, X.M.; Paraschiv, V; Huskens, J.; Reinhoudt, D.N. J. Am. Chem. Soc. 2003, 125, 4279. [ Links ]
[2] Park, S.J.; Taton, T.A.; Mirkin, C.A. Science 2002, 295, 1503. [ Links ]
[3] Bogunia-Kubik, K; Sugisaka, M. Biosystems 2002, 65, 123. [ Links ]
[4] Wieckowski, A.; Savinova, E.R.; Vayenas, C.G. Eds. Catalysis and Electrocatalysis at Nanoparticle Surfaces, Marcel Dekker: New York, 2003. [ Links ]
[5] Blomen, L.J.; Mugerwa, M.N. Eds. Fuel Cell Systems, Plenum Press: New York, 1993. [ Links ]
[6] Zubimendi, J. L.; Vázquez, L.; Ocón, P.; Baró, J.M.; Triaca, W.E.; Salvarezza, R.C.; Arvia, A.J. J. Phys. Chem. 1993, 97, 5095. [ Links ]
[7] Lee, I.; Chan, K.; Phillips, D.L. Appl. Surf. Sci. 1998, 136, 321. [ Links ]
[8] Zoval, J. V.; Lee, J.; Gorer, S.; Penner, R. M. J. Phys. Chem. B 1998, 102, 1166. [ Links ]
[9] Bogozi, A.; Lam, O.; He, H. X.; Li, C. Z.; Tao, N. J.; Nagahara, L.A.; Amlani, J.; Tsui, R. J. Amer. Chem. Soc. 2001, 123, 4585. [ Links ]
[10] Aktary, M.; Lee, C. E.; Xing, Y.; Bergens, S.H.; Mc Dermott, M.T. Langmuir 2000, 16, 5837. [ Links ]
[11] Remita, H.; Keita, B.; Torigoe, K.; Belloni, J.; Nadjo, L. Surf. Sci. 2004, 572, 301. [ Links ]
[12] Atashbar, M.Z.; Bliznyuk, V.; Banerji, D.; Singamaneni, S. J. Alloy Comp.2004, 372, 107. [ Links ]
[13] Walter, E.C.; Zach, M.P.; Favier, F.; Murray, B. J.; Inazu, K.; Hemminger, J.C.; Penner, R.N. Chem. Phys. Phys. Chem. 2003, 4, 131. [ Links ]
[14] Walter, E.C.; Favier, F.; Penner, R.N. Anal. Chem. 2002, 74, 1546. [ Links ]
[15] Rodriguez-Nieto, F.J.; Morante-Catacora, T.Y.; Cabrera, C.R. J. Electroanal. Chem.2004, 571, 15. [ Links ]
[16] Randin, J.T.; Yeager, E. J. Electroanal. Chem. 1975, 58, 317. [ Links ]
[17] Simonov, P.A.; Likholobov, V.A. in Catalysis and Electrocatalysis at Nanoparticle Surfaces, Wieckowski, A.; Savinova, E.R.; Vayenas, C.G. Eds. Marcel Dekker: New York, 2003. [ Links ]
[18] Rice, R. J.; McCreery, R. Anal. Chem. 1989, 61, 1637. [ Links ]

