










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkAnales de la Asociación Química Argentina
Print version ISSN 0365-0375
An. Asoc. Quím. Argent. vol.94 no.1-3 Buenos Aires Jan./July 2006
REVIEW ARTICLES
Molecular recognition and binding mechanism of N-aralkyl substituted 2-aminoindans and the dopamine D2 receptor. A theoretical study
1 Andujar, S. A.; 1Garibotto, F. M.; 1Enriz, R. D.; 2Migliore de Angel, B.; 3Angel-Guío, J.; 4Charris, J.
1 Departamento de Química, Universidad Nacional de San Luis, Chacabuco 917, 5700 San Luis, Argentina. FAX: +54-2652-431301, E-Mail: denriz@unsl.edu.ar.
2 Laboratorio de Síntesis Orgánica y Diseño de Fármacos. Departamento de Química, Facultad de Ciencias. Universidad del Zulia, Apartado N 526, Maracaibo. Venezuela.
3 Sección de Farmacología. Instituto de Investigaciones Clínicas. Dr. Américo Negrette, Facultad de Medicina. Universidad del Zulia, Apartado N 526, Maracaibo. Venezuela.
4 Laboratorio de Síntesis Orgánica. Facultad de Farmacia. Universidad Central de Venezuela. Caracas. Venezuela.
Received March 1st, 2006. In final form April 24th, 2006
Dedicated to Prof. Imre G. Csizmadia on the occasion of his 75th birthday
Abstract
In order to better understand, at submolecular level, the minimal structural requirements for the recognition process in the inhibitor activity, N-aralkylsubstituted 2-aminoindans were examined as D2 dopamine receptor antagonist variants. Semiempirical (AM1) and ab-initio (RHF/3-21G and RHF/6-31G(d)) calculations were performed for a better understanding of the recognition process at submolecular level. Using the above-mentioned computational model, we were able to interpret the basic behavior and predict some additional features of N-aralkyl substituted 2-aminoindans-Dopamine D2 receptor interaction.
Resumen
Se estudiaron N-aralquil-2-substituidos-aminoindanos como variantes de inhibidores del receptor D2 de dopamina con el objeto de mejorar el entendimiento, a nivel molecular, de los requerimientos mínimos estructurales para el proceso de reconocimiento en la actividad inhibitoria. Se utilizaron cálculos semiempiricos (AM1) y ab initio (RHF/3-21G y RHF/6-31G(d)) para entender el proceso de reconocimiento a nivel sub-molecular. Utilizando el mencionado modelo computacional fue posible interpretar el comportamiento básico y predecir algunas características adicionales de la interacción entre los N-aralquil-2-substituidos-aminoindanos y el receptor D2 de dopamina.
Introduction
The brain dopaminergic system has a crucial role in the etiology of several neuropsychiatric disorders, including Parkinson's disease, depression and schizophrenia [1,2].
Since the introduction of chlorpromazine, a known dopamine (DA) antagonist, DA-ergic drugs have been widely used in medicinal practice. Thus, several dopaminergic drugs are used to treat these pathologies, but many problems are attributed to these therapies [1,2]. For these reasons the search for new, more efficient dopaminergic agents with lower adverse effects is still an extremely active research field. Scientific literature in this field is numerous and can be found summarized in relatively recent review articles [3,4].
During the last decades, a large amount of N-substituted 2-aminoindan (compound 2 in Figure 1) and 2-aminotetraline analogs (compound 3) with peripheral and central nervous system action have been reported [5-8]. Recently we reported the synthesis and dopaminergic profile of N-aralkyl substituted (compound 1), which displayed an interesting dopaminergic activity [9]. Now, we are interested to evaluate at submolecular level the minimal structural requirements for the recognition process between compound 1 and its biological receptor. Thus, in the present theoretical study we simulate the electrostatic interactions between compound 1 with its biological receptor in terms of a reduced molecular model. In an attempt to better understand the biological profile reported for compound 1, we present here an exhaustive conformational and electronic study of compound 1. We also discuss a possible molecular recognition and binding mechanism for this compound and the stereoelectronic requirements to elicit the dopaminergic activity.
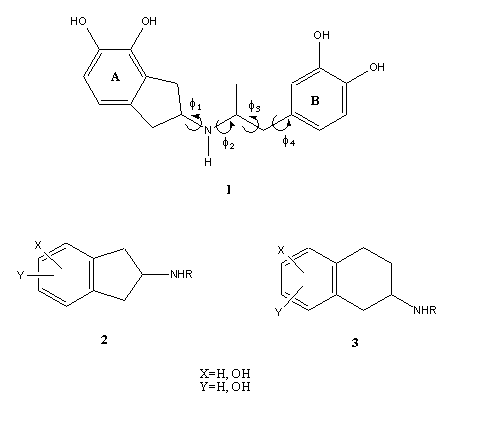
Figure 1. General structure feature of N-substituted 2-aminoindan (2) and 2-aminotetraline analogs (3). Chemical structure of compound 1 showing the different torsional angles (f1,f2, f3 and f4).
Methods of calculation
Conformational and electronic study
To determine the minima on the conformational potential energy hipersurface (PEHS) of compound 1, fully relaxed semiempirical (AM1) and ab-initio (RHF/3-21G and RHF/6-31G(d)) calculations were performed.
An extensive search for low energy conformation on the PEHS of compound 1 was carried out by using 54 input files obtained from multidimensional conformational analysis (MDCA)[10-12] in connection with AM1 semiempirical calculations. Subsequently, several distinct trial atomic spatial arrangements were used in the geometry optimization jobs using ab-initio (RHF/3-21G and RHF/6-31G(d)) calculations to locate the possible equilibrium structures present on the multidimensional energy surfaces. Minima were characterized through harmonic frequency analysis employing RHF/6-31G(d) calculations. Rotational energy profiles around torsional angles have been determined using RHF/3-21G calculations. The energy has been calculated at 30º intervals of the dihedral angles.
All calculations reported here were performed using Gaussian 03 [13]. The electronic study of compound 1 was performed using molecular electrostatic potentials (MEPs). MEPs have been shown to provide reliable information, both on the interaction sites of molecules with point charges and on the comparative reactivities of these sites [14-16]. These MEPs were calculated using RHF/6-31++G(d,p) wave functions. RHF/6-31G(d) coordinates were imported to generate the wave functions; thus, RHF/6-31++G(d,p) single-point calculations were performed from Gaussian 03 program. All MEPs graphical presentations were created using Molekel [17].
Construction of the receptor model
A reduced 3D model of the human dopamine D2 receptor was constructed, based on the theoretical model structure of bacteriorhodopsin [18]. Binding pocket of the D2 DAR (dopamine receptor) was defined according to M. M. Teeter et al. In our reduced model, only 22 aminoacids were included for the molecular simulations. The size of the molecular system simulated and the complexity of the structures under investigation restricted the choice of the quantum mechanical method to be used. Consequently the semiempirical AM1 method [19] was selected. The torsional angles of the ligands and the flexible side-chains of the amino acids as well as the bond angles and bond lengths of the moieties involved in the potential intermolecular interactions were optimized. In contrast, the torsional angles of backbones as well as the bond angles and bond lengths of non-interacting residues were kept frozen during the calculations.
Results and Discussion
Conformational and electronic study of compound 1
The postulate that a molecule has to assume a particular conformation in order to function as a receptor implies that the ability of a molecule to achieve the required conformation would affect its activity. This is usually interpreted in the sense that activity will be impaired unless a molecule can assume the active form. However, it must be pointed out that to relate the biological activity of an antagonist to its conformational properties possess considerable problems since it is difficult to devise molecules of a given conformation without also changing some other physico-chemical properties.
In the first step of our work we performed an exhaustive conformational study of compound 1 because it is essential to know the conformational behavior of this compound in order to understand the drug-receptor interaction; not only in terms of its preferred conformations but also in terms of conformer interconversions. According to Fig 1, the flexible aspects of the molecular conformation of 1 is governed by four torsional angles f1, f2, f3 and f4. Consequently, the potential energy hypersurface (PEHS) of four independent variables
E = f (f1, f2, f3, f4 ) (1)
contains all the conformations as minima. Thus, this compound is, in principle, a quadruple rotor. Multidimensional Conformational Analysis (MDCA) [10-12] predicts the existence of 3 x 3 x 3 x 2 = 54 legitimate minima for a quadruple rotor with these multiplicities. Using MDCA predicted 54 geometries as input, we located a total of 43 conformers on the PEHS (eq. 1) at the AM1 level of theory, instead of the expected 54 structures (Table 1). Next, using the 43 AM1 geometries as input files, we performed RHF/3-21G calculations. Only 33 different conformers were obtained from RHF/3-21G computations (Table 2). To categorize the different structures obtained for this compound, we introduced intuitively rather than by a precise definition, four forms: Fully Folded (FF), Partially Folded (PF), Partially Extended (PE) and Fully Extended (FE). By the FF form, we mean a closed structure with relatively short distances between the aromatic rings. By the FE form, we mean a form with a linearized connecting chain. By PF and PE forms we denote a intermediate conformation between folded and extended. In these two forms the catecol ring can adopt two different spatial orderings: closed in the PF forms and open for the PE conformations.
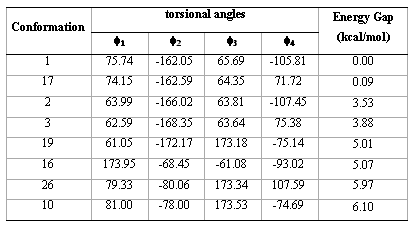
Table 1.Torsional angles and energy gap obtained for the different conformations of compound 1 from AM1 calculations.
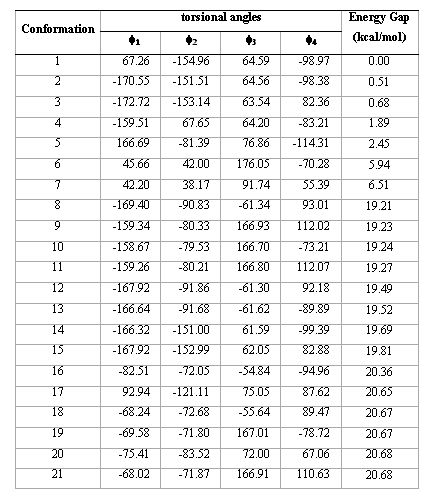
Table1 (continued)
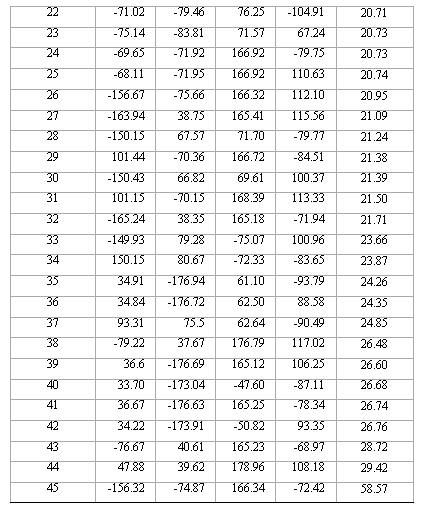
Table 2. Torsional angles and energy gap obtained for the different conformations of compound 1 from RHF/3-21G calculations.
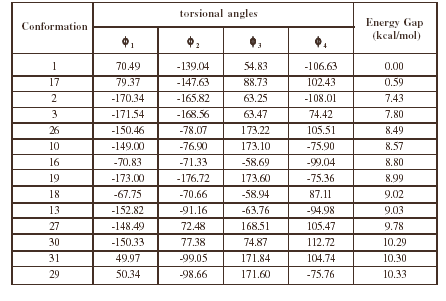
Table2 (continued)

Table 3. Torsional angles and energy gap obtained for the preferred conformations of compound 1 from RHF/6-31G(d) calculations.
The conformational analysis of compound 1 requires at this point the evaluation of its flexibility i.e.; the energy determination of the transitional barriers between the predicted conformers. This is of crucial importance because, if the barriers display low, energy during the molecular recognition this molecule could be converted, with a low energy cost, to a preferred geometry in the binding site within the receptor.
The energy profiles showing the influence of the torsional angles f1 - f4 on the potential energy of the rotamers are given in Figures 2(a) – 2(d), respectively.
The curves of Figures 2(a), 2(b) and 2(c) show that these rotations possess three preferred conformations (gauche +, anti and gauche -). However, the barriers that separate these conformations are somewhat different. The energy barrier for f2 is ≈ 5.5 kcal/mol, whereas the barriers for f1 and f3 are relatively high (≈ 7 kcal/mol) at RHF/3-21G level. Looking at the torsional angle f4 (Fig 3(d)) the preferred conformations were found to be those in which the catecol ring was located perpendicular to the C–C bond (f4≈ ± 90º). The barriers at f4 = 0º and 180º are ≈ 5.5 kcal/mol. This result could explain that practically all the conformations obtained for compound 1 possess the torsional angle f4 in a perpendicular or antiperpendicular position (see Tables 1-3).
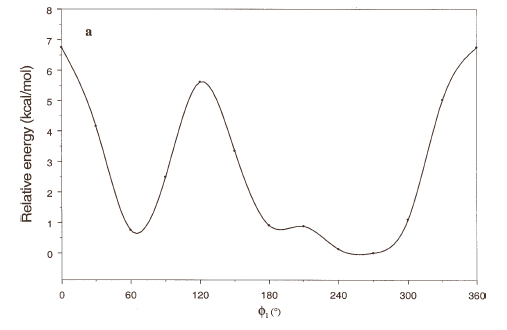
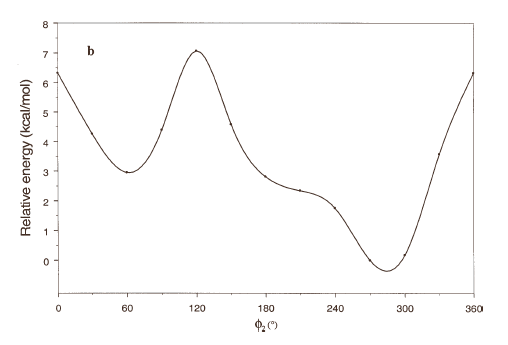
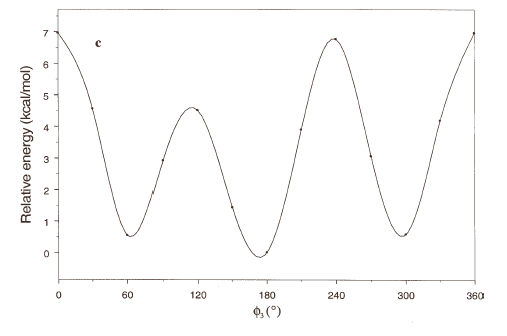
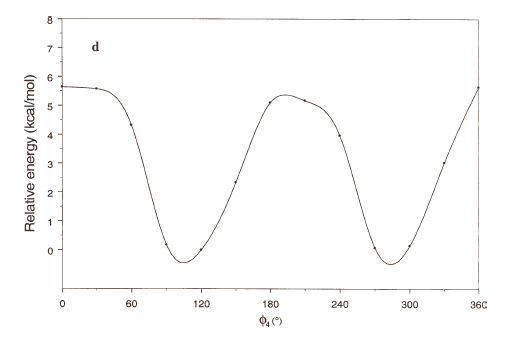
Figure 2. Rotational energy barrier profiles computed at the RHF/3-21G level. a) torsional angle f1, starting with f2, f3 and f4 in -76.00°; 173.00°; -75.00°, respectively. b) Torsional angle f2, starting with f1, f3 and f4 in -150.00°; 173.00°; -75.00°, respectively. c) Torsional angle f3, starting with f1, f2 and f4 in -150.00°; -76.00°; -75.00°, respectively. d) Torsional angle f4, starting with f1, f2 and f3 in -150.00°; -76.00°; 173.00°, respectively.

Figure 3. Spatial view of the preferred low-energy conformations (17, 2, 3, 19, 16, 26 and 10) of compound 1 obtained from RHF/6-31G(d) calculations.
In order to confirm the conformations obtained from RHF/3-21G calculations, we optimize the energetically preferred forms using RHF/6-31G(d) calculations (Table 3); these conformations were confirmed through harmonic frequency analysis.
It is interesting to note that RHF/3-21G calculations found one FE conformation (conformer 19) and one FF form (conformer 16). However, these conformations were not found at RHF/6-31G(d) level. Conformation 19 changes the torsional angle f1 from anti to gauche+ and conformation 16 changes the torsional angle f2 from gauche- to anti; therefore RHF/6-31G(d) calculations predict that all the energetically preferred conformations of compound 1 can adopt PF or PE forms. Also, it is interesting to note that the four preferred forms (conformations 1, 17, 2 and 3) possess a closely related spatial ordering indicating that this type of conformations are highly preferred for compound 1. Figure 3 gives a spatial view for the energetically preferred conformations obtained for compound 1 obtained from RHF/6-31G(d) computations.
The energy differences between the most stable conformation and the other forms, and also the barriers that separate them (predicted by ab-initio calculations) are large enough to suggest that the transition between the different forms is somewhat restricted. Thus, ab-initio calculations predict only a moderate molecular flexibility for compound 1.
Once the preferred conformations of compound 1 were obtained, in an attempt to find the potentially reactive sites for this compound, we evaluate the electronic aspects of this molecule using MEPs. MEPs are of particular value because they permit visualization and assessment of the capacity of a molecule to interact electrostatically with a binding site. MEPs can be interpreted in terms of a stereoelectronic pharmacophore condensing all available information on the electrostatic forces underlying affinity and specificity.
Figure 4 shows the MEPs obtained for compound 1. The MEPs of dopamine is also included in this figure for comparison. It should be noted that both MEPs show a remarkable similarity. In fact the MEPs obtained for compound 1(figure 4a) looks like "two joined dopamine molecules". The close electronic similarity between compound 1 and dopamine could account for their common affinity for the D2-dopamine receptor. This assumption is supported by a qualitative comparison of the isopotential maps obtained through ab-initio calculations.
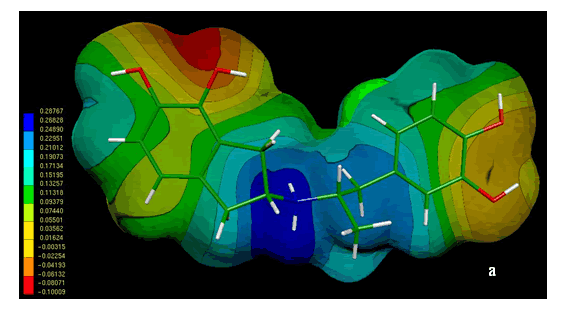
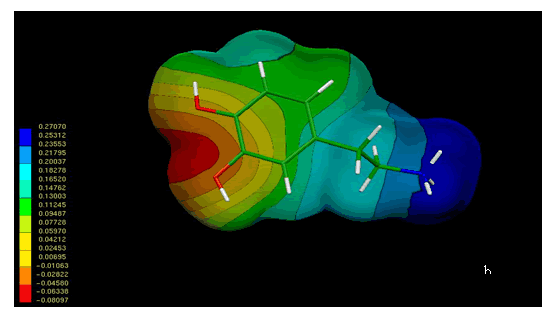
Figure 4. Electrostatic potential-encoded electron density surfaces of the core structures of compounds 1 (a) and dopamine (b). The surfaces were generated with Gaussian 03 after ab-initio minimizations with a 6-31++G(d,p) basis set. The coloring electrostatic potential in red is indicating the strongest attraction to a positive point charge whereas blue is indicating the strongest repulsion. The electrostatic potential is the energy of interaction of the positive point charge with the nuclei and electron of a molecule. It provides a representative measure of overall molecular charge distribution
The analysis of MEPs showed three characteristic regions, two with negative potential (red and orange zones) and another with positive potential (blue zone). The negative regions are generated by the presence of OH groups on the aromatic rings; while the positive region corresponds to the large positive potential around the nitrogen cationic head.
In the case of compound 1, the electrostatic potentials surrounding both aromatic rings displayed an adequate electronic profile to produce electrostatic interactions. The questions which arise are: a) which of the catecol rings of 1 is mimetizing the catecol ring of dopamine and b) which is the role (if any) of the other catecol ring of compound 1. This problem will be discussed in the next section.
A molecular model for the binding mechanism of compound 1
The recent cloning [20-25] of dopamine receptor (D1-D5) suggests that they are members of a large family of G-protein coupled, seven transmembrane receptors [26]. These receptors are similar to the better-characterized rhodopsin protein [27], which spans the plasma membrane seven times, with the transmembrane (TM) domains forming a binding pocket. Based on the amino acid sequence of the dopamine D2-receptor and computer modeling, Sukalovic et al. [28] suggest that dopamine may interact with aspartate86 and the serines (141 and 144). Consistent with the D2 receptor model proposed by Sukalovic et al. [28], the experimental results of Mansour et al. [29] clearly demonstrate the importance of the negatively charged aspartate86 for both dopaminergic agonist and antagonist binding. Elimination of the negative charge by mutating this critical amino acid to either asparagine or glycine markedly reduced the affinity of both agonists and antagonists to the D2 receptors.
The structure of bacteriorhodopsin reported by M. M. Teeter et al. [18] was plotted using the Chimera program [30] (Figure 5). Binding pocket of the D2 DAR was defined according to Sukalovic et al. [28]; while molecular interaction calculations were carried out using semiempirical AM1 computations. The binding pocket used for theoretical calculation is denoted by a circle in Figure 5. The main features of the D2 DAR model shown in Figure 6 using dopamine as the ligand are:
a- salt bridge between protonated nitrogen atom of the flexible side-chain and negatively charged Asp86 (calculated distance 2.34 Å).
b- hydrogen bonds between the OH of catecol ring and Ser144 and Ser141, calculated distance 1.84 and 2.45 Å, respectively.
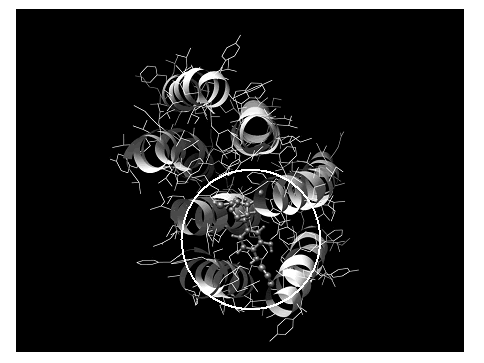
Figure 5. Spatial view of the dopamine D2 receptor model from reference [18] using the Chimera program as graphic interface.

Figure 6. Interactions of dopamine (ligand) with the D2 dopamine receptor. Schematic representation of two interactions: salt bridge to the left and catecol ring with Ser144 and Ser141 to the right. All the heteroatoms involved in the intermolecular interactions are shown in the figure
Our results indicate that the aspartate residue86 serves as a possible anchoring point. Previously, we reported that R-NH+-Asp interactions would be the driving force for the N-alkyl-benzyltethrahydroisoquinolines/D1DAR complex [31]. Those calculations were performed using B3LYP/6-31++G (d,p) calculations and taking into account the solvent effects. Thus it appears that semiempirical AM1 calculations are in agreement with experimental [19] as well as theoretical [31] results using more accurate computations.
Given that the amino group of dopamine is likely anchored at aspartate86, the meta- and para-hydroxyls of the catecol moiety are free to form hydrogen bonds with either serine141 or serine144. The calculated interatomic distances for these interactions are shown in Figure 6.
Examination of the structure of compound 1 and computer modeling of the D2 receptor suggest that unlike dopamine, compound 1 could form hydrogen bonds with serine144 using ring A or ring B. Both situations are shown in Figures 7 and 8, respectively.

Figure 7. Interactions of compound 1 (ligand) with the D2 dopamine receptor. Schematic representation of the salt bridge (down), the catecol ring A with Ser144 and Ser141 to the right and catecol ring B with His189 and Phe185 (denoted by a circle). All the heteroatoms involved in the intermolecular interactions are shown in the figure.

Figure 8. Interactions of compound 1 (ligand) with the D2 dopamine receptor. Schematic representation of the salt bridge (down), the catecol ring B with Ser144 and Ser141 to the right and catecol ring A with His189 and Phe185 (denoted by a circle). All the heteroatoms involved in the intermolecular interactions are shown in the figure.
Figure 7 gives a spatial view of the molecular interaction between compound 1 and D2 DA receptor in which the ring A is interacting with Ser144 and Ser141. The salt bridge between protonated N of the flexible connecting chain and the negatively charged Asp86 displayed a distance of 2.69 Å, whereas the hydrogen bonds between the OH of catecol ring A and Ser144 and Ser141, displayed a calculated distance of 1.68 and 2.63 Å, respectively. The total energy of this complex is –0.896605 hartree.
Figure 8 gives a spatial view of the molecular interaction between compound 1 and D2 DA receptor in which the ring B is interacting with Ser144 and Ser141. In this case, the salt bridge between protonated N of the flexible connecting chain and the negatively charged Asp86 displayed a distance of 3.73 Å, whereas the hydrogen bonds between the OH of catecol ring A with Ser144 and Ser141 , displayed a calculated distance of 2.14 and 2.78 Å, respectively. The total energy of this complex is –0.825268 hartree. Thus, the energy gap between these complexes is 44.76 Kcal/mol. On the other hand, the interatomic distances obtained in the first complex are shorter than those attained in the second one. These results indicate that the complex in which catecol ring A is interacting with both serines is energetically preferred with respect to the other complex.
Finally, we wish to discuss some details about the role in compound 1 of the catecol ring which is not interacting with Ser144 and Ser141 (ring B). It is clear that in order to obtain a clear profile of the overall recognition process it is necessary to underline the role of this moiety in the binding mechanism of compound 1. If it is assumed, as it seems highly probable, the ligand will have only a single conformation in its complex with the receptor; thus, the conformation of this complex must involve a process of conformational selection (or alternatively, the ability of the ligand to achieve the required conformation) which will influence the kinetics and energetic of complex formation.
In principle, at least three different roles might be attributed to catecol ring B, i.e:
(i) conformational restriction
(ii) steric hindrance
(iii) a third possibility is that this ring could contribute to its own interaction through a van der Waals interaction
Although these concepts are independently formulated, they might be interdependent.
Our theoretical calculations predict that His189 in TM VI as well as Phe185 could be also involved in van der Waals interactions (zone denoted with a circle in Figure 6). Thus, AM1 computations indicate that the catecol ring B is positioned in the ancillary pocket spanned by two conserved aromatic residues, i.e Phe185 and His189 (Figure 9). It appears that an additional hydrogen bond can be built with His189. However, more accurate calculations considering the electronic correlation are required to confirm this assumption.
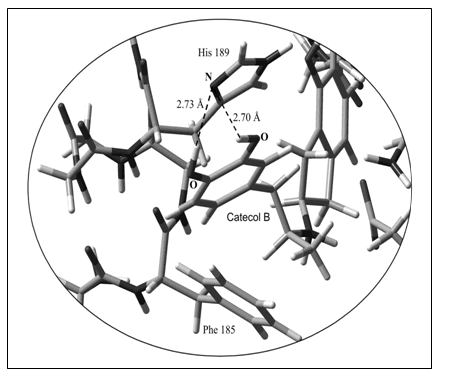
Figure 9. Spatial view of the molecular interaction of catecol ring B with the ancillary pocket (His189 and Phe185). Non-interacting atoms have been deleted in this figure to better appreciate the intermolecular interactions. All the heteroatoms involved in the intermolecular interactions are shown in the figure.
The results of AM1 calculations presented here show that the molecular mechanism for the recognition process of the dopamine D2-receptor is energetically feasible. The general picture is that both the salt bridge interaction and the hydrogen bonds between the catecol ring A with Ser144 and Ser141 play a key role in the molecular recognition process. In addition, an appropriate ring orientation of the other catecol moiety (ring B) may be operative stabilizing this process. Thus, a kind of stepwise binding involving first the salt bridge followed by the rest of the molecule seems a reasonable possibility and there is some strongly suggestive evidence that this phenomenon takes place.
Conclusions
To better understand how N-aralkylsubstituted 2-aminoindans interact with dopamine D2 receptors, the conformational and electronic properties of compound 1 have been studied using ab-initio calculations. A putative binding mechanism of compound 1 to dopamine D2 receptor has been prepared on theoretical calculations grounds. The results of molecular interaction simulations on compound 1-D2DAR complexes revealed that this ligand interacts through protonated N of the connecting flexible chain with Asp86 (salt bridge) as well as hydrogen bonds between one of the catecol (ring A) of the ligand with Ser144 and Ser141. In addition, an appropriate ring orientation of the other catecol moiety (ring B) may be operative stabilizing this process. The results presented here provide a basis for further rational design of DA-ergic compounds structurally related to N-aralkylsubstituted 2-aminoindans.
Acknowledgements
This work was supported by grants from the National University of San Luis, Argentina. R.D. Enriz is a member of CONICET-Argentina. S.A. Andujar thanks CONICET and F.M. Garibotto thanks ANPCyT for Research Fellowships.
References
[1] Baldesarini, R. J. Drugs and treatment of psychiatric disorders: Psychosis and anxiety. In: Hardman, J. G., Limbird,L.E. (Eds), The Pharmacological Basis of Therapeutics, McGraw-Hill: New York, 1996. [ Links ]
[2] Stahl, P. Essential Psychopharmacology. Neuroscientific Basis and Practical Applications. 2nd Edn.; Cambridge University: New York, NY, 2000 [ Links ]
[3] Ananth, J.; Burgoyne, K.S.; Gadasalli, R.; Aquino, S. J. Psychiatry Neurosci. 2001, 26, 385. [ Links ]
[4] Remington, G. J. Psychiatry Neurosci. 2003, 28, 275 [ Links ]
[5] Cannon, J.G.; Kim, J. C.; Aleem, M. A. J. Med. Chem. 1972, 15, 348Links ] Times, serif">.
[6] Cannon, J.G.; Pérez, J.A.; Pease, J.P. J. Med. Chem. 1980, 23, 745. [ Links ]
[7] Cannon, J.G.; Pérez, J.A.; Bhatnager, R.K.; Long, J.P.; Sharabi, F.M. J. Med. Chem. 1982, 25, 1442. [ Links ]
[8] McDermed, J.D.; McKenzie, G.M.; Phillips, A.P. J. Med Chem. 1975, 18, 362. [ Links ]
[9] Angel-Guío, J.E.; Migliore de Angel, B.; Suarez-Roca, H.; Israel, A.; Charris, J.E.; Enriz, R.D. Sintesis y evaluacion farmacologica de nuevos agentes aralquilicos con actividad dopaminergica central, XXVI Meeting of the Argentine Chemical Society, September 2006, pp 81. [ Links ]
[10] Csizmadia, I.G., The Chemistry of the thiol group, John Wiley and sons, New York, 1974, Chap 1. [ Links ]
[11] Peterson M.P.; Csizmadia, I.G. J. An. Chem. Soc. 1978, 100, 6911. [ Links ]
[12] Peterson, M.P.; Csizmadia, I.G., Progress of theoretical organic chemistry, Vol 3, Elsevier, Amsterdam, 1982, pp. 190-266. [ Links ]
[13] Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C. and Pople, J. A. Gaussian 03, Revision B.05 2003, Gaussian, Inc., Pittsburgh PA. [ Links ]
[14] Polilzer, P.; Truhlar, D.G. Chemical Applications of Atomic and Molecular Electrostatic Potential, Plenum, New York, 1991. [ Links ]
[15] Carrupt, P.A.; El Tayar, N.; Karlé, A.; Festa, B. Methods Enzymol 1991, 202, 638. [ Links ]
[16] Greeling, P.; Langenaeker, W.; De Proft, F.; Baeten, A. Theoretical and Computational Chemistry. 1996, 3, 587. [ Links ]
[17] Flükiger, P.; Lüthi, H.P.; Portmann, S.; Weber, J MOLEKEL 4.0; Swiss Center for Scientific Computing: Manno, Switzerland, 2000. [ Links ]
[18] Teeter, M.M.; Froimowitz, M.F.; Stec, B.; Durand ,C.J. J. Med. Chem. 1994, 37, 2874 [ Links ]
[19] Dewar, M.S.; Zoesbisch, E.G.; Healy, E.F.; Stewart, J.P. J. Am. Chem. Soc. 1985, 107, 3902. [ Links ]
[20] Bunzow, J.R.; Van Tol, H.H.; Grandy, D.K.; Albert, P.; Salon, J.; Chistrie, M.; Machida, C.A.; Neve, K.A.; Civelli, O. Nature. 1998, 336, 783. [ Links ]
[21] Dearry, A.; Gingrich, J.A.; Falardeau, P.; Fremeau, R.T. Jr.; Bates, M.D.; Caron, M.G. Nature. 1990, 347, 72. [ Links ]
[22] Mahan, L.C.; Burch, R.M.; Monsma, F.J.; Sibley, D.R. Proc. Natl. Acad. Sci. 1990, 87, 2196. [ Links ]
[23] Sokoloff, P.; Giros, B.; Martes, M.P.; Bouthenet, M.L.; Schwartz, J.C. Nature. 1990, 347, 146. [ Links ]
[24] Sunahara, R.K.; Guan, H.C.; O`Dowd, B.F.; Seeman, P.; Laurier, L.G.; Ng, G.; George, S.R.; Torchia, J.; Van Tol, H.H.M.; Niznik, H.B. Nature. 1991, 350, 614. [ Links ]
[25] Van Tol, H.H.M.; Bunzow, J.R.; Guan, H.C.; Sunahara, R.K.; Seeman, P.; Niznik, H.B.; Civelli, O. Nature. 1991, 350, 610. [ Links ]
[26] Gilman. A.G. Annu. Rev. Biochem. 1987, 56, 615. [ Links ]
[27] Henderson, R.; Balwin, J.M.; Ceska, T.A.; Zemlin, F.; Beckman, E.; Downing, K. J. Mol. Biol. 1990, 213, 899. [ Links ]
[28] Sukolovic, V.; Andric, D.; Roglic, G.; Kostic-Rajacic, S.; Schrattenholz, A.; Soskic, V. European Journal of Medicinal Chemistry. 2005, 40, 481. [ Links ]
[29] Manzour, A.; Meng, F.; Meador-Woodruff, J.H.; Taylor, L.P.; Civelli, O.; Akil, H. European Journal of Pharmacology- Molecular Pharmacology Section. 1992, 227, 205. [ Links ]
[30] Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. J. Comput. Chem. 2004, 25, 1605. [ Links ]
[31] Suvire, F.D.; Cabedo, N.; Chagraoui, A.; Zamora, M.A.; Cortez, D.; Enriz, R.D. Theochem. 2003, 666, 455. [ Links ]

