










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Asociación Química Argentina
versión impresa ISSN 0365-0375
An. Asoc. Quím. Argent. v.94 n.1-3 Buenos Aires ene./jul. 2006
REGULAR PAPERS
Theoretical study of the outer valence photoelectron spectra of methyl nitrite and fluoromethyl nitrite
1Estrada, M. R.;1 Zamarbide, G. N. ;2Sánchez-Marín,*J.
1 Universidad Nacional de San Luis. Chacabuco y Pedernera 5700, San Luis, Argentina
2 Universitat de València, Institut de Ciència Molecular, Apartado de Correos 22085. Valencia E-46071. Spain
FAX: +34 96 354 3274; E-Mail: jose.sanchez@uv.es
Received March 31st, 2006. In final form May 2nd, 2006
Dedicated to Prof. Imre G. Csizmadia on the occasion of his 75th birthday
Abstract
The effect of the syn-anti conformational isomerism and fluorination of methyl nitrite on the ultraviolet photoelectron spectra of methyl nitrite is predicted on the basis of Outer Valence Green Function calculations using the 6-311++G** basis set. The conformational space of methyl nitrite and its single, di- and tri-fluoromethyl derivatives have been explored with MP2 and B3LYP-DFT approaches. Only the fluoro-methyl nitrite shows minima with geometries compatible with bound systems, therefore the OVGF study has been limited to methyl nitrite and fluoro-methyl nitrite. A procedure has been devised to represent simulated spectra starting from the OVGF data and the experimental UPS spectrum taken from the literature. The He I experimental spectrum has been assigned at the OVGF level and the simulated spectra are discussed. It is predicted that the syn-anti conformational isomerism will affect less the spectrum of the fluorinated system than that of the methyl nitrite. The effects of fluorination will be larger for the second peak than for the first. Fluorination brings about changes of the general shape (e.g., peak shifts, changes in gap widths, positions, etc.) of the low ionization energies of the spectrum.
Resumen
Se predice el efecto del isomerismo conformacional syn-anti y de la fluoración del nitrito de metilo sobre el espectro fotoelectrónico ultravioleta del nitrito de metilo a partir de cálculos con el método de funciones de Green para la valencia externa (OVGF) usando el conjunto de base 6-311++G**. El espacio conformacional del nitrito de metilo y sus derivados uni, di y tri-sustituidos se ha explorado con las aproximaciones MP2 y B3LYP-DFT. Unicamente el nitrito de fluormetilo muestra mínimos que tienen geometrías compatibles con sistemas enlazados. Por esa razón el estudio OVGF se ha limitado al nitrito de metilo y a su derivado monofluorado. Se ha puesto a punto un procedimiento para representar los espectros simulados a partir de los datos proporcionados por el cálculo OVGF y el espectro experimental del nitrito de metilo tomado de la literatura. El espectro He I experimental ha sido asignado de acuerdo con los resultados OVGF, y esto ha permitido discutir los espectros simulados a partir de los cálculos. Se predice que el isomerismo conformacional syn-anti afectará menos al espectro del derivado fluorado que al del nitrito de metilo. El efecto de la substitución por F será mayor en el segundo pico del espectro que en el primero. Los cambios en el espectro producidos por la fluoración serán fácilmente reconocibles por los notables cambios en la forma general del espectro (desplazamiento de los picos, cambios en las separaciones entre bandas, etc.) en la región de bajas energías de ionización.
Introduction
The reactivity of Alkyl nitrites, and notably that of the first member of the series, methyl nitrite (MeONO), has deserved interest because these compounds are sources of the methoxy and NO radicals [1,2,3]. The properties of methyl nitrite [4] and other alkyl nitrites [5] as nitrosation agent have been also recently studied. On the other hand, the cis-trans (more properly, syn-anti) unimolecular isomerization with respect to the O-N single bond of methyl nitrite (see scheme 1) has been the subject of accurate theoretical and experimental studies because in this case a low energy barrier occurs, along with a low density of vibrational states [6].

The relative stability of syn and anti conformers in alkyl nitrites has also been studied in detail [7,8]. In particular, for methyl nitrite, the syn conformer appears as the most stable for a wide range of temperatures [7], while for ethyl and higher alkyl nitrites, the situation is more involved [8,9] and stereoelectronic effects, dynamic chirality and other conformational effects related to the cis-trans behaviour with respect to the C-O bond need to be considered.
Flourinated alkyl nitrites seem to have been little studied and references to fluorinated alkyl nitrites are hard to find. Fluorine is a strong electronegative substituent that increases the electron density in its proximity. It is well known that CF3- is a prototype of meta-orienting with deactivation substituent in aromatic substitutions that leads to 100% meta-oriented nitration in fluoromethyl-benzene [10]. On the other hand, the nitrooxymethane group is very reactive as indicated above. One can think of fluorinated alkyl nitrites as reactive and unstable. In fact, as it will be shown in the results section, stability difficulties related to fluorination of the methyl nitrite can be expected. Hence, the theoretical study of the electronic properties of fluorinated alkyl nitrites appears to be in order as experimental studies will face difficulties. It can be advanced here that, as it will be shown in the results section, only the singly fluorinated methyl nitrite is found to show minima with common geometrical properties.
This preliminary study is focussed on the one-electron energies that are commonly measured by photoelectron spectroscopy and related techniques. At this point we can take benefit of the experimental UPS spectrum of methyl-nitrite reported by D. Wang et al. [11]. Ultraviolet photoelectron spectroscopy (UPS) is an appealing technique to theoreticians as it can be interpreted on the basis of the MO theory provided by Koopmans theorem [12]. However, it is well known that the correlation and orbital relaxation effects not included in the one-electron Hartree-Fock model are needed for a proper accounting of the ionization energies measured through the UPS techniques [13,14]. This is generally true even for those cases where the ionization process can be properly described as a single-particle single-step process. The outer valence Green function (OVGF) method [15,16,17,18] provides a convenient way for taking into account correlation and relaxation effects in a direct (i.e., not as a difference of energies) calculation of ionization potentials (IP), conserving the description of the ionization process as related to a single MO.
This work aims to calculate the UPS spectra of methyl nitrite and fluoro-methyl nitrite by means of the OVGF method in order to evaluate the influence of the syn-anti conformations in the UPS spectra and the influence of F substitution in the UPS spectrum of methyl nitrite. Another goal of this work is to use the experimental UPS spectrum of methyl nitrite as a pattern against which the calculated data can be compared. In order to get realistic simulated spectra the calculation was started from the available experimental data of the UPS spectrum of methyl nitrite [11]. The procedure is described in the next section.
Methods and Calculation Conditions.
The vertical one-electron ionization potentials, IP, have been calculated with the SCF and the OVGF methods. Geometry optimizations have been performed with the DFT approach with the B3LYP functional (Becke hybrid three-parameter nonlocal exchange functional combined with the Lee-Yang-Parr dynamic correlation one) [19,20]. As shown below, the DFT results reveal that, for the fluorinated methyl nitrites, methods to calculate explicitly the dynamic correlation effects are required. As a consequence, the optimized MP2 geometries have been preferred to the DFT in the OVGF calculations.
All the calculations reported in this work have been obtained with the valence triple zeta, polarized and singly augmented basis set usually denoted as 6-311++G(d,p). This basis set provides a convenient compromise between the desired accuracy (suitable for the purposes of the present work) and the required computational effort, both for geometry exploration and OVGF ionization energies.
Previous calculations on the conformational space of ethyl and methyl nitrites can be found in the literature [6,8,9]. Taking the existing works on alkyl nitrites as a guide, the set of expected conformers of methyl nitrite and fluorine substituted methyl nitrite have been optimized for the conditions of the present work. Such optimization of geometries has been attempted using the B3LYP density functional. The non-substituted and single-fluorinated molecules have led to well conditioned and geometrically consistent minima at the B3LYP level. However, doubly and fully substituted methyl nitrites have led to minima that show abnormally long single bond O-N length (see the results section for further details).
At long bond distances, the van der Waals dispersion effects between the two resulting molecular fragments (the [NO.]-like fragment and the [O-CF3.]-like fragment) can be critical in order to describe properly the molecular system. Unfortunatelly, the DFT models cannot deal in most cases with inter-molecular or inter-fragment dispersion effects [21]. These effects require a detailed calculation of simultaneous local single excitations that can be accounted for, e.g., by MP [22] approaches of second order (at least). Hence, all the geometrical structures in the present work have been optimized looking for MP2 energy minima. It is clear that in those cases where the minima show conventional bond distances and angles, the B3LYP geometry and energies should be considered better. However, in the present case a consistent set of energies and geometries can be more likely obtained with the MP2 approach in order to deal with inter-fragment dispersion effects. All the minima have been verified analytically to have all positive hessian eigenvalues.
As it was indicated above, the outer valence IP's corresponding to each structure have been calculated with the OVGF approach [15,16,17]. The OVGF method can be described as a third-order (with extensions in which some higher order terms are taken into account) perturbative-type single particle Green's function method [18]. These kind of methods have as zero order the Koopmans' ionization potentials, so that a one to one correspondence can be established between each OVGF pole and just one MO from which the electron is removed.
The UPS spectra have been simulated by means of gaussian curves weighted by the OVGF pole strength squares and centered at the IP values. The FWHM was 0.5 eV. throughout. This FWHM value accounts for the non-resolved vibrational widening of the low IP bands in ordinary low resolution spectra. This simple procedure cannot reproduce some effects, such as the spread of intensities of each one-electron ionization in a number of multi-electron processes (e.g. shake-up lines), that attenuate the intensities and crowd the lines at high IP values. In order to reproduce the experimental attenuation of the spectra of alkyl-nitrites as reported by Wang et al. [11], the whole calculated spectrum has been dumped by a single gaussian centered at the onset of the spectrum. The exponent of this dumping gaussian factor (-0.02) was chosen to simulate the attenuation of the low resolution experimental spectrum of methyl nitrite [11]. This experimental spectrum is conveniently simulated by centering each band at the experimental band maximum reported in ref. [11], and giving the same weight (0.8) and same FWHM (0.5 eV) to each gaussian curve. The same dumping gaussian factor has then been used in all the calculated spectra. However, in the case of calculated spectra, the individual contribution of each band has been weighted by the respective squared pole strength provided by the OVGF calculation. All the MP2, DFT and OVGF calculations reported in this work have been performed with the gaussian 03 program system [23].
Results
Table 1 shows the energies of the minima at the SCF, MP2 and B3LYP-DFT levels. Only the energy values of the most stable conformations for each syn and anti conformer are reported. Other conformations having the formal properties of minima can be found for the fluorinated compounds, but they are not reported for the sake of simplicity. The distances of the single O-N bond in Å are also in table 1. The SCF minima show a consistent trend towards a moderate increasing of the O-N single bond length with the number of F atoms. However, both DFT and MP2 results predict uncommonly long O-N equilibrium distances. For the F-methyl nitrite conformers, the value got for the O-N bond length is about 1.5 Å at B3LYP and MP2 levels. The O-N bond length values are less similar in the CF2H- and CF3- cases. As discussed above, in the methods section, at this point the authors prefer to rely in the MP2 geometries and energies. As a consequence, the study of the single-electron outer valence ionization energies of two- and three- fluorinated methyl nitrites has been discarded. It can be of some interest to note that, contrarily to what happens with methyl nitrite, in the fluoro-methyl case the anti conformer is found to be more stable than the syn counterpart.
Table 1 Energy (hartree) and O-NO single bond distance for the SCF, MP2 and B3LYP minima (6-311++G(d,p) basis set) of methyl nitrite and fluorinated methyl nitrites.

The IP values according to the Koopmans' theorem and OVGF calculation are reported for anti conformers of methyl and fluoro-methyl nitrite in table 2. The corresponding values for the syn conformers are reported in table 3.
Both tables 2 and 3 include a column with pictorial schemes of the orbitals from which the electron are removed in each one-electron ionization process whose energy is given. These figures can help the reader to figure out the distribution of signs and nodal surfaces around the (essentially planar) C-O-N=O structure. In general, the studied molecular structures have as a whole C1 symmetry, but it can be helpful to distinguish between p-like and s-like MO's according to their behaviour relative to the C-O-N=O plane.
Table 2. Molecular orbitals and vertical ionization energies (eV) for the anti conformers of methyl and fluoro-methyl nitrite.
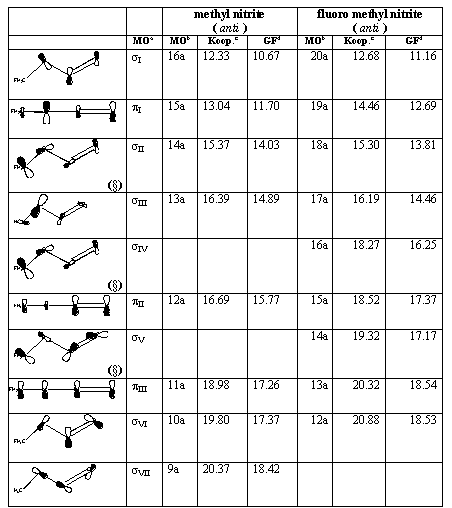
(§) MO's with significant p-like electron density centered in the F atom.
a MO's denoted according to their s-like or p-like behaviour. Notation for this work. See text for details.
b MO's denoted according to the C1 point group symmetry.
c Koopmans' IP values (6-311++G(d,p) basis set). This work.
d OVGF IP values (6-311++G(d,p) basis set). This work.
Table 3. Molecular orbitals and vertical ionization energies (eV) for the syn conformers of methyl and fluoro-methyl nitrite.

(§) MO's with significant p-like electron density centered in the F atom.
a MO's denoted according to their s-like or p-like behaviour. Notation for the present work. See text for details.
b MO's denoted according to C1 point symmetry group.
c Koopmans' IP values (6-311++G(d,p) basis set). This work.
d OVGF IP values (6-311++G(d,p) basis set). This work.
The HOMO is denoted as sI . The other MO's classified as s are denoted as sII, sIII , and so on, accordingly to their increasing Koopmans' energies. A similar notation is used for p-like MO's. This notation makes easier the task of comparing the results for non-substituted and F-substituted systems, as well as the assignation of the spectra features. The conventional notation based on the C1 (non)symmetry group has been also provided in tables 2 and 3 so that the correspondence of each MO and spectrum peaks with the new notation is simple to set no matter the molecule, the calculation level and the structure under consideration.
Only the occupied MO's that lead to converged poles in the OVGF calculations are reported in tables 2 and 3. All the MO's reported are consecutive in the canonical one-electron energy values (Koopmans). Note, however, that in some cases the order of the ionization energies is changed by the OVGF calculation (see, e.g., the pII and sV MO's in table 2 and the pII and sIII MO's in table 3).
The simulation of the experimental He I -UPS spectrum reported by Wang et al. [11], as it results from the procedure described in the methods section, has been included in figure 1. The same figure shows the calculated simulation of the UPS spectra of non-substituted methyl-nitrite for the syn and anti forms.
The bands of the simulated spectra shown in figure 1 have been labelled A to G. The OVGF assignment of the bands can be obtained from tables 2, 3 and 4 with the help of these labels and the ad hoc notation described above. The assignment on the basis of Koopmans' theorem is not shown for simplicity, but it can be easily established from tables 2 and 3.
The main features of the experimental spectrum [11] can be described as follows (see table 4). 1) The onset of the spectrum is sharp and close to 10.0 eV. 2) The two first bands at 11.03 and 11.83 eV are well resolved. 3) A separated band at 13.50 eV occurs after a gap of ˜1 eV. 4) After the second gap a broad band shows two maxima at 14.75 and 15.44 eV. 5) Two more separated bands of decreasing intensity occur at about 16.8 and 18.5 eV, the last band was not assigned by the authors in reference [11].
All these features are essentially present in the simulation of the experimental spectrum shown in figure 1, even if the last two bands appear as too much intense. All these features are present also in the simulations based in the OVGF results of the syn conformer. They are also present in great part in the OVGF-based simulation for the anti conformer.
The greatest discrepancy between the experimental and the calculated spectra occurs in the intensity of the peak labelled as F. The double intensity of the F peaks in the OVGF spectra is a consequence of the occurrence of two IP values that cannot be resolved under the conditions of the spectra simulation.
Table 4. Descriptive assignment of the main features in the experimental UPS spectrum of methyl nitrite and the calculated UPS spectra. See text for details.

a Spectra features. See figures 1 and 2.
b Assignment and experimental IP values (eV, in parenthesis) as reported by D. Wang et al. in ref [11].
c D and E peaks appear as unresolved for the simulation conditions in the syn case of methyl nitrite.
d A low intensity signal at 18.5 eV appears in the experimental spectrum in ref. [11] that was not assigned by the authors.

Figure 1.Simulated UPS spectra of methyl nitrite according to the procedure described in the text and using data from the experimental spectrum[11] and OVGF calculations (this work)
The predicted spectra for the two conformers of the fluoro-methyl substituted nitrite are shown in figure 2. The simulated spectrum of methyl-nitrite (based on the experimental data) is shown in figure 2 as it was in figure 1, in order to make comparisons easier. The bands in the spectra of the fluorinated systems are labelled A* to G*. In this way, the effects on the spectrum due to fluorination are easily shown and compared to the A to G features in figure 1. These effects are summarized in table 5. The following facts deserve to be pointed out:
1) The outer bands appear displaced towards higher IP's . However, the p-like B* band appears more displaced than the s-like A* band. As a consequence, a deep gap is predicted between the A* and the B* peaks. Note that the B* peak occurs in the region (12.0-13.0 eV) where the first large gap occurred in the spectrum of the non substituted molecule.
2) The bands C* and D* appear closer to each other than the bands C and D in methyl nitrite. The data in table 5 show that this effect is mainly due to the large displacement of the D* band towards smaller IP's in both syn and anti conformers. Note, however, that even for the conformer the gap between C* and D* features is smaller as compared to that between C and D.
3) As a consequence of points 1) and 2), the A* to D* peaks set gathers in the range 10-15 eV, while the A-D peaks of methyl nitrite extends from 10-16 eV
4) Another consequence of the large shift of D* is the prediction of a great gap between the D* and the E* band. Another way of seeing this is to say that, as a consequence of the points above, the gap between the outer valence and the inner valence parts of the spectrum becomes larger. Note also that the E peak appears as unresolved in the syn case in figure 1.
5) The F* and G* features show smaller displacement. These features are predicted too much intense, in particular for the anti conformers where unresolved peaks are obtained.
Discussion
The errors expected from OVGF calculations should be taken into account in order to discuss the results shown in the section above. For the outermost vertical valence ionization potentials, in the case of molecules having second row atoms (Li-Ne), errors of ±0.2 eV can be expected with the OVGF method [18]. Our basis set does not include f functions, so that we will assume larger errors, say ±0.3 eV [18]. These values are of the same order than the FWHM that we have used for the simulation (0.5 eV).

Figure 2. Simulated UPS spectra of fluoro-methyl nitrite according to the procedure described in the text and using data from the OVGF calculations (this work)
Errors in the inner valence region of the spectra can be significantly larger. In fact, the features concerning the highest IP values do not deserve a detailed discussion because the OVGF predictions, made on the basis of one-electron processes, are much less reliable as one approaches the inner valence region of spectra. Hence, the discussion is focused on the A-D (A*-D*) features of the spectra. All these features, but the B (B*) one (see table 4) can be assigned. Note, however, that the E (E*) feature can be assigned as p-like in the methyl nitrite forms, but as s-like in the fluoro-methyl case.
OVGF and SCF results
It is worth mentioning that a one to one correspondence exists between the MO energies and the OVGF values. The correlation effects decrease the four IP's assigned to A-D (A*-D*) peaks by about 1.4 eV in the methyl-nitrite and about 1.6 eV in the mono substituted forms. Under the conditions of the spectra simulations, this change is about three times the FWHM of the bands. These changes fall in the range from 1.27 to 1.77 eV. Their relevance is not only quantitative because the order of the assignments can change, as it happens for the pII MO of the syn form of methyl-nitrite. It becomes clear that the correlation effects cannot be left out for a realistic simulation of the spectra.
Table 5. Effect of F substitution in the calculated UPS spectra of methyl nitrite as given by the shift in the OVGF ionization potential values (eV).

a See figures 1 and 2.
b mn stands for methyl nitrite
c fmn stands for fluoro-metyl nitrite
d Averaged from -0.44 and -0.96 eV
e Averaged from -0.13 and -0.25 eV
Syn and anti forms
As reported in table 1, the syn conformer appears as the most stable form of methyl-nitrite, while the anti form is found more stable in the fluoro-methyl nitrite.
The differences between the syn and anti simulations show up in figures 1 and 2. In the flourinated molecule, only the A* and the C*-D* peaks are significantly affected by the syn-anti isomerism. Therefore, apart from the shift of the first peak (that implies the shift of the spectrum onset), the only region affected by the conformational changes around the O-N single bond would be that in the range of 13.5 - 15 eV.
The differences between the syn and anti spectra are more relevant for methyl nitrite, in this case, all the peaks are shifted by 0.12-0.35 eV (absolute values). In particular, the C and E features are the only shifted towards smaller IP values, and their shift amount to -0.23 and -0.26 eV respectively.
Note that the first IP, corresponding to the A and A* features in figures 1 and 2, is incremented by ca. 0.2 eV in the syn forms, no matter if the molecule is fluorinated or not. However, the B* peak, assigned to the p-like MO closer to the HOMO, is not affected. This behaviour is predicted by the Koopmans' approach, and confirmed by the correlated OVGF results.
F substitution effects
The changes and general features introduced in the spectrum by the presence of the F atom have been described in detail in the previous section. We will only say here that they are relevant enough to assure that the F-methyl nitrite spectrum can be easily discriminated from that of the non substituted systems. The shift of the B* band by 1.0 eV towards the region of the first deep gap in the methyl nitrite spectrum at 12.5 eV can be a key feature to describe the effect of F substitution.
Conclusions
The OVGF results place properly the onset of the spectrum and its general shape, even if a overall dumping function has been used to simulate the intensity attenuation at large IP values. The changes related to the syn-anti conformerism can be significant, although taking it into account appears to be less relevant in the fluorinated case than in methyl-nitrite one. The changes in the spectrum due to the replacement of an H by a F atom in the methyl nitrite are predicted to be relevant, both in shifts as in the general shape of the spectrum.
The present calculations have been performed with a limited basis set, but the effects related to the F substitution are in most cases larger than the expected errors. However, the effects related to the syn-anti conformerism were found to be smaller than the expected errors. These results could be refined by using larger basis sets that include at least f functions, as the expected errors could, in that case, become smaller that ±0.2.
Acknowledgments
This research was supported by the Spanish MEC, Spanish GV and European FEDER Funds, (Projects CTQ2004-07768-C02/BQU and GV-INFRA03-047). M.R.E. thanks the spanish GV for a visitor grant (AINV06/069).
References
[1] Steacie, E.W.P.; Show, G.T. Proc. Roy. Soc., Ser. A, 1934, A146, 388 [ Links ]
[2] He, Y; Sanders, W. A.; Lin, M. C. J. Phys. Chem. 1988, 92, 5474 [ Links ]
[3] Fernandez-Ramos, A.; Matinez-Nuñez, E.; Rios, M. A., Rodriguez-Otero, J.; Vazquez, S. A.; Eztevez, C. M.; J. Am. Chem. Soc. , 1998, 120, 7594. [ Links ]
[4] Niiya, T.; Ikeda, H.; Yukawa, M.; Goto Y.; Chem. Pharm. Bull., 2002, 50, 1502 [ Links ]
[5] Ikeda, H.; Yukawa, M.; Niiya, T., Chem. Pharm. Bull. 2005, 53, 820. [ Links ]
[6] Martinez-Nuñez,E.; Vazquez, S. A., J. Chem. Phys. 1997, 107, 5393. [ Links ]
[7] Conboy, C. B.; Chauvel, J. P.; Moreno, P. O.; True, N. S.; Ott, C. M. J. Phys. Chem. 1986, 90, 4353. [ Links ]
[8] Suter, H. U.; Nonella, M.; J. Phys. Chem. A, 1997, 101, 5580 [ Links ]
[9] Bombasaro, J.A.; Suvire, F.D.; Chasse, G.A.; Zamarbide, G.N.; Estrada, M.R. J.Molec. Struct. THEOCHEM 2001, 548, 39. [ Links ]
[10] Roberts, J. D.; Caserio, M.C., Basic Principles of Organic Chemistry, Benjamin, 1965, New York. [ Links ]
[11] Wang, D.; Li S.; Li Y.; Zheng S.; Ding C.; Gao Y.; Chen W. J. Electron Spec. Rel. Phen. 1996, 82, 19. [ Links ]
[12] Koopmans A. Physica, 1933, 1, 104. [ Links ]
[13] Eland, J. H. D.; Photoelectron Spectroscopy, 2nd. edition. Butterworths. London, 1984. [ Links ]
[14] Hüfner, S. Photoelectron Spectroscopy, 2nd. edition. Springer-Verlag, Berlin, 1995. [ Links ]
[15] Cederbaum, L.S.; J. Phys. B 1975, 8, 290. [ Links ]
[16] Ortiz, J.V. J. Chem. Phys. 1988, 89, 6348. [ Links ]
[17] Zakrzewski,V. G.; Ortiz, J. V. Int. J. Quantum Chem. 1995, 53, 583. [ Links ]
[18] Yeager, D.L; "Multiconfigurational Green's function (propagator) techniques for excitation energies, ionization potentials and electron affinities: an overview" in Mukherjee D., Editor, "Applied Many-Body Methods in Spectroscopy and Electronic Structure". Plenum Press, New York, 1992., 133-161. [ Links ]
[19] Lee, C.; Yang, W.; Parr R. Phys. Rev. B, 1988, 37, 785. [ Links ]
[20] Becke, A.D. .J. Chem Phys. 1993, 98, 5648. [ Links ]
[21] Cramer, C.J. Essentials of Computational Chemistry. 2nd. edition. Wiley. Chichester. 2004. [ Links ]
[22] Moeller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 359. [ Links ]
[23] Gaussian 03, Revision C.02, Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Montgomery; Jr. J. A.; Vreven T.; Kudin K. N.; Burant J. C.; Millam J. M.; Iyengar S. S.; Tomasi J.; Barone V.; Mennucci B.; Cossi M.; Scalmani G.; Rega N.; Petersson G. A.; Nakatsuji H.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Klene M.; Li X.; Knox J. E.; Hratchian H. P.; Cross J. B.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Ayala P. Y.; Morokuma K.; Voth G. A.; Salvador P.; Dannenberg J. J.; Zakrzewski V. G.; Dapprich S.; Daniels A. D.; Strain M. C.; Farkas O.; Malick D. K.; Rabuck A. D.; Raghavachari K.; Foresman J. B.; Ortiz J. V.; Cui Q.; Baboul A. G.; Clifford S.; Cioslowski J.; Stefanov B. B.; Liu G.; Liashenko A.; Piskorz P.; Komaromi I.; Martin R. L.; Fox D. J.; Keith T.; Al-Laham M. A.; Peng C. Y.; Nanayakkara A.; Challacombe M.; Gill P. M. W.; Johnson B.; Chen W.; Wong M. W.; Gonzalez C.; and Pople J. A.; Gaussian, Inc., Wallingford CT, 2004. [ Links ]

