










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkSalud(i)Ciencia
Print version ISSN 1667-8682On-line version ISSN 1667-8990
Salud(i)ciencia vol.23 no.1 Ciudad autonoma de Buenos Aires June 2018
REVISIONES
Mutación en el gen TMEM70: una forma de aciduria 3-metilglutacónica con fenotipo variable. Presentación de dos casos clínicos
Mutation in the TMEM70 gene: a form of 3-methylglutaconic aciduria with variable phenotype. Presentation of two clinical cases
Gonzalo Armani 1, María Cruz Tubio 1, Ana Clara Bernal 1, Hernán Diego Eiroa 1
1 Hospital de Pediatría SAMIC Prof Dr. Juan P. Garrahan, Ciudad de Buenos Aires, Argentina
Recibido: 18-06-2018
Aceptado: 22-06-2018
Resumen
La excreción urinaria de ácido 3-metilglutacónico es el sello distintivo de un conjunto de entidades en las que la disfunción mitocondrial es el eje cardinal fisiopatológico. A lo largo de los años, a modo didáctico, se han utilizado distintas nomenclaturas para agrupar fenotípica, genética o bioquímicamente un creciente número de pacientes en los que el ácido 3-metilglutacónico se pone de manifiesto en la corrida de ácidos orgánicos urinarios. Al reconocer dichos patrones fenotípicos y bioquímicos, los pediatras, neurólogos, genetistas y especialistas en enfermedades metabólicas pueden caracterizar de manera más certera los distintos subgrupos que componen estas enfermedades, entre los cuales las mutaciones en el gen TMEM70 pertenecen a un subtipo de características fenotípicas y bioquímicas en constante descubrimiento. Es nuestra intención aportar la experiencia de dos pacientes con diagnóstico de aciduria 3-metilglutacónica con mutación en el gen TMEM70, seguidos en el Hospital Juan P. Garrahan con el fin de poder difundir el conocimiento de este tipo de enfermedades, para, de esta forma, aumentar su sospecha clínica entre la comunidad médica.
Abstract
The urinary excretion of 3-methylglutaconic acid is the hallmark of a series of clinical entities whose mitochondrial dysfunction is the physiopathological cardinal sign. Over the years, in teaching, many nomenclatures have been used to group, in a phenotypic, genetic or biochemical manner, an increasing number of patients in whom 3-methylglutaconic acid can be found in urinary organic acid tests. Pediatricians, neurologists, geneticists and specialists in inborn errors of metabolism can better characterize the different subgroups that comprise these pathologies when they identify the different phenotypic and biochemical patterns. Within these subgroups, the TMEM70 gene variant belongs to a subtype with phenotypic and biochemical characteristics that are constantly being discovered. We will contribute the experience of two patients with 3-methylglutaconic aciduria with a TMEM70 gene variant, followed in the Juan P. Garrahan Children's Hospital, in order to spread the knowledge of this type of diseases, and therefore enhance its clinical detection in the health community.
Introducción
La aciduria 3-metilglutacónica (3MGA-uria) es un grupo heterogéneo de patologías que incluye distintos errores congénitos del metabolismo, bioquímicamente caracterizados por la excreción de ácido 3-metilglutacónico (3MGA) y ácido 3-metilglutárico (3MG).1 El 3-metilglutaconil CoA (3MG-CoA), precursor del 3MGA, es un intermediario que se forma durante el catabolismo de la leucina, un aminoácido ramificado.1 2 En la orina de individuos sanos el ácido 3MGA puede encontrarse en forma de trazas (menos de 20 mmol/mol creatinina), mientras que en los pacientes con 3MG-uria las concentraciones pueden elevarse (aun en forma intermitente) por encima de 1000 mmol/mol creatinina.1 3 El catabolismo completo de la leucina en la mitocondria produce acetil-CoA, que luego se oxida en el ciclo del ácido cítrico para la formación de adenosín-trifosfato (ATP) generando de esta manera energía.2 Es interesante destacar que el 3-metilglutaconil CoA se encuentra asociado de manera creciente a un número de errores congénitos en los que se presenta un compromiso del metabolismo energético mitocondrial.2
Sólo en la deficiencia de la 3-metilglutaconil CoA hidratasa es conocido el origen del 3MGA, por lo que puede ser considerada como una 3MGA-uria primaria. En contraste con los restantes fenotipos en los que se manifiesta la 3MGA-uria, la patogenia de la excreción del 3MGA no está del todo clara. Existen distintos modelos que intentan explicar su formación: vía el "shunt del mevalonato", en el cual está involucrada la síntesis de colesterol, o secundario a un desequilibrio energético a nivel mitocondrial, donde la acumulación de acetil-CoA finalizaría en la producción de niveles aumentados de 3MGA.2
La 3MGA-uria es el sello de un grupo de enfermedades fenotípicamente heterogéneas que han sido históricamente catalogadas con números romanos: I (aciduria primaria OMIM ID: #250950), II (síndrome de Barth, TAZ, OMIM ID: #302060), III (síndrome de Costeff, OPA3, OMIM ID: #258501) y V (síndrome de miocardiopatía dilatada con ataxia [sus siglas en inglés: DCMA], DNAJC19 OMIM ID: #610198).3 Además de estos síndromes bien definidos, existe un creciente grupo de pacientes "sin clasificar" que presentan una marcada excreción de 3MGA y que se han englobado en el grupo IV (OMIM ID: #25951), es en este subgrupo en el cual las mutaciones del gen TMEM70 se encuentran incluidas. El gen TMEM70 codifica para la proteína encargada de la estabilidad y ensamblaje de la denominada ATP sintetasa, también conocida como complejo V de la cadena de fosforilación oxidativa, cuya función final es la producción de energía en forma de ATP.4 6 La mutación del gen TMEM70 es a su vez el defecto más común codificado por el ADN nuclear que afecta la ATP sintetasa.6 Actualmente, y según una nueva clasificación, los pacientes son agrupados desde un punto de vista genético3 (Tabla 1).
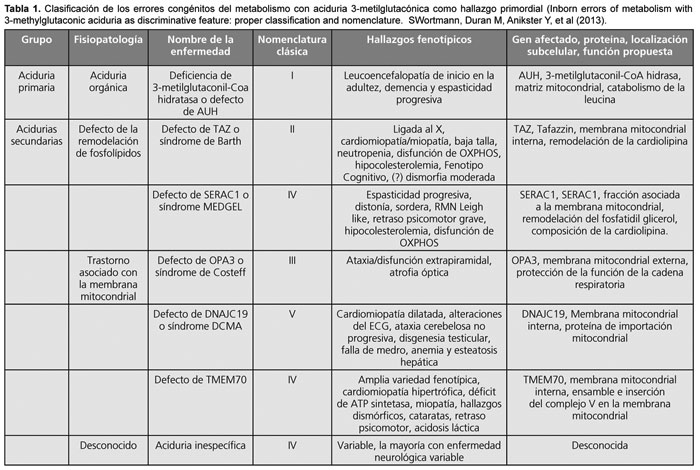
Como se puede observar en la Tabla 1, diversas características fenotípicas hacen posible el encuadre de los distintos subtipos, es así que la presentación de inicio en la adultez (grupo I), la asociación de cardiopatía y neutropenia (grupo II), la atrofia óptica (grupo III) y la ataxia cerebelosa no progresiva (grupo V) son hallazgos distintivos que permitirán al equipo médico establecer un diagnóstico clínico. En contraste, las anormalidades fenotípicas y bioquímicas del grupo IV son mucho más variables respecto de los otros subgrupos, dificultando de esta manera su encuadre. Teniendo en cuenta que la descripción más grande de casos de mutación en el gen TMEM70 comunicada hasta el momento no supera los 48 pacientes,6 consideramos importante contribuir con dos casos que, presentando mutaciones en este gen, han demostrado una evolución diferente en algunos aspectos clásicamente descriptos hasta el momento.
Caso 1
Se trata de una niña de 9 años, de origen romaní, nacida de 34 semanas con un peso de 1410 g (por debajo del percentilo 3 de peso para la edad), con diagnóstico prenatal de retraso de crecimiento intrauterino (RCIU), secundario a hipertensión materna. Hija de padres consanguíneos (Fgura 1). El puntaje de Apgar fue 6/8. Tanto la pesquisa neonatal, el fondo de ojo, como también las otoemisiones acústicas fueron normales. La niña permaneció ocho semanas internada en neonatología por presentar hiperbilirrubinemia, (luminoterapia por dos días), síndrome de distrés respiratorio y sepsis intrahospitalaria sin rescate de germen. Durante esta internación se realiza ecografía abdominal y cerebral, ambas normales.
A los 5 meses de vida presenta una intercurrencia infecciosa con shock y deshidratación, por lo que requiere internación en unidad de cuidados intensivos (UCI) por 16 días. Al año de vida se evalúa su antropometría y su desarrollo neuromadurativo, con los siguientes puntajes Z: peso -3.9; talla -3.4, y perímetro cefálico -4, con retraso madurativo marcado. Por este motivo se realiza una valoración multidisciplinaria y se determina que la paciente, además de los hallazgos clínicos antes mencionados, presenta: acidosis metabólica con GAP aumentado, hiperuricemia (9.7 mg/dl) e hiperlactacidemia (11.3 mmol/l); se realiza ecografía abdominal en la que se identifican riñones disminuidos de tamaño. El ecocardiograma informa: aorta bicúspide, insuficiencia aórtica leve a moderada, leve hipertrofia de ventrículo izquierdo, sin dilatación de cavidades.
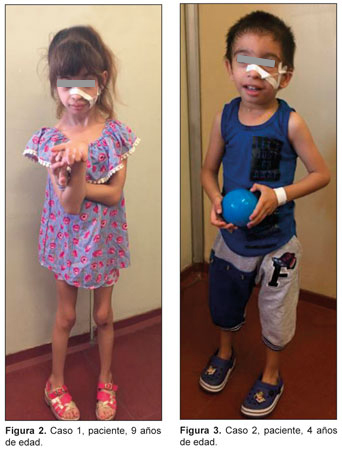
A los 13 meses es derivada al Hospital Juan P. Garrahan para completar estudios. Dado que la paciente persiste con hiperuricemia (10.2 mg/dl) e hiperlactacidemia (59.6 mg/dl) es evaluada por el Servicio de Errores Congénitos del Metabolismo. La niña presenta un fenotipo peculiar similar a los ya descriptos en este subgrupo, con paladar ojival, frente amplia, epicanto con telecanto, pelo ralo, mentón triangular, pectus excavatum, pie bot y hernia inguinal1 5 (Figura 1), como también un retraso de crecimiento grave, caracterizado por puntajes Z de peso -3.53, talla -3.21, y perímetro cefálico -3.8.
Como parte de la valoración se solicitan ácidos orgánicos urinarios que evidencian excreción leve de ácido 3-metilglutárico y moderada de ácido 3-metilglutacónico. En el análisis de aminoácidos plasmáticos se observa una elevación de los niveles de alanina: 68 µmol/dl (VN 14 a 45 µmol/dl) y de glicina 43 µmol/dl (VN 10 a 34.3 µmol/dl).
Con estos hallazgos se sospecha el diagnóstico de aciduria 3-metilglutacónica con fuerte sospecha del subtipo tipo IV con mutación en el gen TMEM70, dada la asociación de excreción en orina de ácido 3-metilglutárico y 3-metilglutacónico, hiperlactacidemia, dismorfias, miocardiopatía, retraso madurativo y de crecimiento. Dicho diagnóstico se confirma a sus 8 años de edad mediante la realización de la secuenciación del gen TMEM70, realizada en el Istituto Neurologico Carlo Besta, de Milán, Italia, por la Dra. Eleonora Lamantea, en la que se halla la mutación homocigota c.317-2A>G en el intrón 2.
Respeto de la función renal, la paciente evoluciona hacia la enfermedad renal crónica en estadio II, asociada con una tubulopatía perdedora de bicarbonato, con necesidad de suplementación. A sus 8 años y 3 meses se realiza una ecografía abdominorrenal que informa riñones con aumento de ecogenicidad sin diferenciación corticomedular.
La paciente presenta a lo largo de su evolución un claro retraso en la adquisición de pautas motoras: sostén cefálico a los ocho meses, se sienta al año, gatea al año y dos meses y deambula a los dos años y dos meses. A los 7 años se realizó evaluación formal del desarrollo y se constata nivel cognitivo disminuido según lo esperado para la edad, correspondiente a una discapacidad intelectual no especificada (escala de inteligencia Stanford-Binet: puntaje fuera de tabla), y a los 8 años comienza a asistir a una escuela de educación especial de manera intermitente.
A los 9 años y tres meses logra deambular sola, dice palabras aisladas, responde a su nombre, no controla esfínteres y no logra tomar objetos. Se alimenta con sonda nasogástrica (SNG) por presentar trastornos deglutorios y manifestar aversión alimentaria.
En lo concerniente al aspecto oftalmológico, presenta esotropía intermitente, ptosis leve de ojo derecho, con motilidad y fondo de ojo normales.
En cuanto al tratamiento, entre los 2 y 7 años, ante la sospecha de una mitocondriopatía recibió biotina, cinc, coenzima Q y vitaminas A, D y C; dicho tratamiento que no demostró mejoría alguna en su evolución y fue suspendido tras la confirmación molecular. Hasta su último control la paciente, de 9 años y 3 meses, continúa presentando marcada hiperlactacidemia (media 44.52 mg/dl; rango 25 mg/dl a 111.5mg/dl). Cabe destacar que no ha presentado cuadros de descompensación metabólica, como tampoco hiperamoniemia, condiciones ampliamente descriptas en esta patología.6
Caso 2
Se trata de un niño de 4 años de origen romaní, hermano de la paciente del caso 1. Se informa diagnóstico prenatal de RCIU. Nace a las 39 semanas, peso de 1200 g (por debajo del percentilo 3 para edad) y perímetro cefálico de 26 cm (por debajo del percentilo 3 para edad), presenta criptorquidia con ambos testículos en orificio inguinal interno bilateral. Puntaje de Apgar 8/9. Pesquisa neonatal normal. Permaneció internado dos meses en neonatología para recuperación nutricional, durante la cual presentó hipoglucemia, acidosis metabólica e hiperlactacidemia. Desde ese momento se encuentra suplementado con bicarbonato.
Por los antecedentes familiares a los tres meses de vida se solicita análisis de ácidos orgánicos urinarios, que solo muestran excreción moderada de ácido láctico, sin detectarse 3MGA. En la determinación de aminoácidos que encuentran niveles de alanina de 412 µmol/l (VN < 345 µmol/l). Presenta hiperlactacidemia persistente (media: 68.6 mg/dl; rango 36 mg/dl a 105 mg/dl) e hiperuricemia secundaria.
Desde el punto de vista cardiológico, tiene un ecocardiograma realizado en el período neonatal donde se manifiesta hipertrofia ventricular izquierda con buena función ventricular. Dicho estudio se repite a los 11 meses de vida y se agrega estenosis supravalvular aórtica, con aorta bicúspide y dilatación de la aorta descendente. En su última evaluación, a los 3 años de vida, evolucionó a insuficiencia aórtica leve.
El paciente presenta enfermedad renal crónica en estadio II desde los 21 meses de vida, con proteinuria de 0.397 g/l (VN < 0.12 g/l), asociada con una ecografía renal que muestra discreta disminución de la diferenciación corticomedular.
En lo concerniente al fondo de ojo se observó disminución del brillo macular y dispersión pigmentaria a los 9 meses de vida.
Respecto del neurodesarrollo, a los tres meses el paciente manifestaba un aumento del tono global, ángulo poplíteo de 90 grados bilateral y reflejo cocleopalpebral negativo. A los 2 años y 3 meses presentaba un temblor fino generalizado, daba pasos con apoyo y decía cuatro palabras. Se realiza evaluación formal del desarrollo a los 2 años y 11 meses, informando: CAT 11.5 meses, cociente 50 y CLAMS 13 meses, cociente de 56. A los 3 años y 3 meses el paciente es capaz de deambular sin apoyo y no controla esfínteres. Presenta a su vez hipoacusia moderada con probable compromiso neurosensorial; al igual que su hermana, tiene un marcado retraso crónico de crecimiento.
El paciente se alimenta con SNG por tener videodeglución patológica para líquidos desde los 9 meses y para sólidos desde los 15 meses.
Dada la gran similitud de hallazgos entre el paciente y su hermana, a pesar de no haber presentado en los ácidos orgánicos urinarios excreción de 3MGA, se decide secuenciar el gen TMEM70 y se encuentra la misma mutación homocigota c.317-2A>G en el intrón 2.
Discusión
La 3MG-uria, tanto primaria como secundaria, agrupa en sí misma un creciente y variado grupo de fenotipos clínicos que pueden remedar otras entidades genéticas pero que presentan como sello distintivo la excreción de 3MGA en la corrida de ácidos orgánicos urinarios, puede ser intermitente o estar presente solo en algún momento de la evolución del paciente.1 3
Es importante destacar que independientemente del subgrupo en el cual se encuadre este tipo de pacientes, la asociación de cardiomiopatía, retraso psicomotor, dismorfias faciales, hiperlactacidemia, hiperamoniemia y compromiso renal deben hacer sospechar al pediatra clínico una enfermedad metabólica. Si bien en primer lugar esta podrá ser englobada como enfermedad con compromiso mitocondrial, y como tal, sometida al puntaje de Nijmegen,7 entre los estudios a realizar de forma obligatoria se encuentra la corrida de ácidos orgánicos urinarios. Teniendo en cuenta que la incidencia particular de cada una de las distintas patologías de índole mitocondrial es baja, la prevalencia global de algún tipo de mitocondriopatía se encuentra en 1/5000 en la población general,8 razón por la cual el conocimiento de la existencia de estas entidades es importante a la hora de considerar los diagnósticos diferenciales de síndromes multisistémicos. La presencia de 3MGA en la orina será tanto un biomarcador de disfunción mitocondrial, como también una clave para el encuadre clínico y posterior estudio molecular de este grupo de trastornos. Desde la descripción de las mutaciones en el gen TMEM70 la asociación ha sido establecida con la etnia romaní,4 en la cual por motivos culturales la consanguineidad tiene una alta incidencia, por lo que según un modelo mendeliano de herencia génica aumenta la probabilidad de que una enfermedad de carácter recesivo se manifieste. Aun así, cabe destacar también que se han comunicado casos de pacientes de etnia no romaní, por lo que actualmente esta enfermedad se considera panétnica.5 6 9
La existencia de crisis metabólicas acompañadas de hiperamoniemia, las cuales pueden generar y agravar el estado neurológico de los pacientes se encuentran en la bibliografía.6 Se ha postulado como causa secundaria de la hiperamoniemia una disminución en la síntesis de carbamoilfosfato y de ácido arginino succínico, ambos componentes del ciclo de la urea, secundarios a la disfunción mitocondrial propia de la enfermedad.4 Es interesante resaltar que entre las manifestaciones que se pueden describir en la descompensación metabólica, distintas de la hiperamoniemia, podemos encontrar un empeoramiento de los parámetros bioquímicos (hiperlactacidemia, hiperalaninemia, hiponatremia e hiperuricemia) sin un aumento significativo de la excreción de 3MGA.6 En nuestra descripción solo el caso índice manifestó una descompensación a los 5 meses de vida, que llevó a la realización de los estudios diagnósticos, aun así, la niña no ha presentado ningún episodio de hiperamoniemia hasta la actualidad.
El compromiso renal, descripto en hasta un 34% de los pacientes con mutaciones en el gen TMEM70,6 arroja algo de luz en la aun desconocida prevalencia del compromiso renal en las citopatías mitocondriales.8 Particularmente en el caso de las mutaciones del gen TMEM70, la patología renal puede manifestarse como acidosis tubular, hidronefrosis o insuficiencia renal aguda/crónica. No es de sorprender que el compromiso renal se manifieste en este tipo de enfermedades, dada la riqueza de mitocondrias que presentan ciertas áreas del riñón como el túbulo contorneado proximal.10
Los pacientes presentados han manifestado a lo largo de su evolución distintos grados de compromiso nefrológico, hasta llegar a una enfermedad renal crónica de tipo II sin que medien otros indicadores diferenciales para asociar sus componentes nefrológicos a un peor resultado evolutivo.
En cuanto al tratamiento de este grupo de enfermedades, en particular la asociada con la mutación del gen TMEM70, la ausencia de una terapia curativa hace que el objetivo de las medidas terapéuticas sea el sostén clínico del paciente, asegurando una correcta alimentación para evitar las descompensaciones metabólicas.6
Conclusión
Las 3MGA-urias son enfermedades multisistémicas y engloban un gran número de pacientes que pueden presentarse con manifestaciones clínicas heterogéneas, muchas de las cuales son frecuentes y conocidas por los pediatras. En la era de los estudios genéticos creemos que una evaluación clínica completa por parte del pediatra y un análisis riguroso por parte del equipo de laboratorio podrá orientar a las familias hacia la mejor opción de estudios moleculares, evitando prácticas que podrían no ser necesarias (por ejemplo, biopsia muscular) para el encuadre del paciente. Nuestro objetivo es destacar la posibilidad de diagnosticar adecuadamente las enfermedades metabólicas en pacientes que se presenten con afección de múltiples órganos asociada con alteraciones bioquímicas y características fenotípicas compatibles.
Consideramos fundamental el trabajo interdisciplinario entre las distintas áreas, las más relevantes son: nefrología, neurología, cardiología, especialistas en metabolismo y bioquímicos.
Esperamos que estas descripciones fenotípicas ayuden en un futuro en el diagnóstico de otros casos y que abran un nuevo grupo de posibilidades en el diagnóstico diferencial de un no menor número de pacientes sin diagnóstico etiológico.
Bibliografía
1. Wortmann S, Kluijtmans L, Engelke U, et al. The 3-methylglutaconic acidurias: what's new? J Inherit Metab Dis 35:13-22, 2010. [ Links ]
2. Ikon N, Ryan R. On the origin of 3 methylglutaconic acid in disorders of mitocondrial energy metabolism. J Inherit Metab Dis 39:749-756, 2016. [ Links ]
3. Wortmann S, Duran M, Anikster Y, et al. Inborn errors of metabolism with 3-methylglutaconic aciduria as discriminative feature: proper classification and nomenclature. J Inherit Metab Dis, 2013. DOI 10.1007/s10545-012-9580-0. [ Links ]
4. Shchelochkov O, Li F, Wang J, et al. Milder clinical course of type IV 3-methylglutaconic aciduria due a novel mutation in TMEM70. Mol Genet Metab 101:282-285, 2010. [ Links ]
5. Torraco A, Verrigni D, Rizza T, et al. TMEM70: a mutational hot spot in nuclear ATP synthase deficiency with a pivotal role in complex V biogénesis. Neurogenetics, 2012. DOI 10.1007/s10048-012-0343-8. [ Links ]
6. Magner M, Dvorakova V, Tesarova M. TMEM70 deficiency: Long-term outcome of 48 patients. J Inherit Metab Dis, 2014. DOI: 10.1007/s10545-014-9774-8. [ Links ]
7. Wolf N, Smeitink J. Mitochondrial disorders: a proposal for consensus diagnostic criteria in infants and children. Neurology 59(9):1402-5, 2002. [ Links ]
8. Emma F, Montini G, Parikh S, et al. Mitochondrial dysfuction in inherited renal disease and acute kidney injury. Nature Reviews, 2016. DOI:10.1038/nrneph.2015.214. [ Links ]
9. Atay Z, Bereket A, Turan S, et al. A novel homozygous TMEM70 mutation resuts in congenital cataract and neonatal mitocondrial encephalo-cardiomyopathy. Gene 515:197-199, 2013. [ Links ]
10. Emma F, Montini G, Salviati L, et al. Renal mitochondrial cytopathies. Internat J Nephro, 2011. DOI: 10.4061/2011/609213. [ Links ]

