










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAgriscientia
versión On-line ISSN 1668-298X
Agriscientia vol.39 no.1 Córdoba jun. 2022
Artículos
Validación de agrupamientos para representar estructura genética poblacional
Cluster validation to depict popula-tion genetic structure.
M. E. Videla 1
C. Bruno 1
1 Videla, M. E. (ORCID 0000-0002-6286-992X), Universidad Nacional de Córdoba, Facultad de Ciencias Agropecuarias, Estadística y Biome-tría. Unidad de Fitopatología y Modelización Agrícola, Consejo Nacional de Investigaciones Científicas y Tecnológicas (UFyMA -CONICET). Universidad Nacional de Villa María, Argentina. Bruno,C. (ORCID 0000-0002-3674-7128), Universidad Nacional de Córdoba, Facultad de Ciencias Agropecuarias, Estadística y Biometría. Unidad de Fitopatología y Modelización Agrícola, Consejo Nacional de Investigaciones Científicas y Tecnológicas (UFyMA -CONICET).
Correspondencia a: eugeniavidela12@gmail.com
DOI: 10.31047/1668.298x.v39.n1.34015
Fecha de recepción: 20/07/2021
fecha de aceptación: 14/12/2021
RESUMEN
Desde los comienzos de la estadística, ha existido la necesidad de identificar el número subyacente de grupos existentes en una población, para dar respuestas a genetistas con respecto a la estructura que se forma por similitudes entre individuos de una o más poblaciones. Se han propuesto numerosos índices para obtener el número óptimo de grupos, que conforman la estructura genética poblacional (EGP). Sin embargo, no hay consenso sobre cuáles son los de mejor desempeño. Para determinar el número óptimo de grupos que definen la EGP, se realizó un estudio de simulación de nueve escenarios de EGP con tres números de subpoblaciones (k = 2, 5 y 10) y tres niveles de diferenciación genética, recreando varios genomas de maíz, para evaluar cuatro índices de validación internos: CH, Connectivity, Dunn y Silhouette. En este estudio, se encontró que los índices de Dunn y Silhouette tuvieron el mejor desempeño para identificar el verdadero número de grupos subyacentes; mientras que Conectividad, el peor. Este estudio ofrece una alternativa sólida para revelar la EGP existente, facilitando así los estudios de población y las estrategias de mejoramiento de cultivos. Además, los presentes hallazgos pueden tener implicaciones para otras especies.
Palabras clave: datos genéticos, análisis de conglomerados, índices de selección, análisis exploratorios de datos.
SUMMARY
Since the beginning of statistics, the identification of the underlying number of existing groups in a population has been a research question aimed at answering geneticists regarding the structure that is formed by similarities between individuals of one or more populations. Numerous indices have been proposed to obtain the optimal number of groups that make up the population genetic structure (PGS). However, there is no consensus on which are the best. In order to determine the optimal number of groups constituting the PGS, a simulation study was conducted of nine PGS scenarios with three subpopulation numbers (k = 2, 5, and 10) and three levels of genetic differentiation recreating various maize genomes to evaluate four internal validation indices: CH, Connectivity, Dunn and Silhouette. This study found that the Dunn and Silhouette indices had the best performance in identifying the true number of underlying groups while Connectivity had the worst. This study offers a robust alternative to unveil the existing PGS, thereby facilitating population studies and breeding strategies in maize programs. Moreover, the present findings may have implications for other crop species.
Keywords: genetic data, cluster analysis, index selection, exploratory data analysis.
INTRODUCCION
El análisis de conglomerados ha demostrado ser una herramienta eficiente para identificar el agrupamiento natural o subyacente de los genotipos de una población (Peña-Malavera et al., 2014). Varios algoritmos han sido desarrollados para clasificar genotipos, dentro de subpoblaciones, utilizando datos genéticos provenientes del genotipado molecular (Lawson et al., 2018; Lee y Tracy, 2009; Odong et al., 2011).Los algoritmos de conglomerado no supervisado son aquellos que no demandan información previa sobre los grupos en los que se espera que los individuos o genotipos se clasifiquen. Por el contrario, los algoritmos denominados supervisados asumen que, los individuos, tienen una asignación a un grupo conocida previamente (Eick et al., 2006).
Uno de los principales desafíos en el análisis de conglomerados es que, dado que los algoritmos de agrupamiento no supervisados definen grupos que no son conocidos apriori, independientemente del método de agrupamiento aplicado, la partición final de los datos requiere alguna clase de evaluación (Rezaee et al., 1998). Es por ello que, los agrupamientos obtenidos por dichos métodos, ya sean supervisados o no, se complementan con cálculos estadísticos desarrollados para estimar, y validar, la cantidad de conglomerados que subyacen en la estructura de los datos poblacionales, y que son de interés identificar (Balzarini et al., 2011). El procedimiento que evalúa el resultado del agrupamiento es conocido como validación del agrupamiento, y tiene como finalidad confirmar si la partición de las observaciones, o el agrupamiento final obtenido, es el que mejor representa a la estructura subyacente de los datos (Charrad et al., 2014; HalkidiyIordanis, 2011). Es importante destacar que, diferentes algoritmos de agrupamiento sobre un mismo conjunto de datos producen distintas configuraciones de unión de los individuos. Por ello, la evaluación de la efectividad en la clasificación y el criterio de agrupamiento seleccionado son críticos, para tener confianza en los resultados de los agrupamientos. Al mismo tiempo, ningún método de agrupamiento indica, de manera automática, el número de grupos encontrado en el conjunto de datos sometido a la clasificación. Esto último genera, en el investigador o usuario del método, el dilema del número óptimo de grupos a seleccionar, como resultado final del método de agrupamiento. Numerosos índices han sido propuestos para dar respuesta al número óptimo de grupos (Charrad et al., 2014). Estos índices trabajan combinando información acerca de la compactación intra-grupo y el aislamiento inter-grupos, así como otros factores que se encuentran relacionados a la geometría y propiedades estadísticas de los datos, el número de observaciones y la medida de similaridad/ disimililaridad utilizada para conformar la matriz de distancia, a partir de la cual trabajan los algoritmos de agrupamiento. Los índices de validación interna utilizan información del conjunto de datos como, por ejemplo, la matriz de proximidad, sin considerar información adicional para seleccionar el número de subpoblaciones (Halkidi et al., 2000). Investigadores de diversas disciplinas han propuesto distintos criterios para la selección de número óptimo de grupos. Así, el índice de Dunn fue propuesto en el área de matemática aplicada (Dunn, 1974) y el estadístico H, en computación estadística (Hartigan, 1975). Más recientemente, se ha propuesto el método L en datos computacionales (Salvador y Chan, 2004). En el área de la biología, se propusieron índices como el CH, que fueron aplicados sobre datos bacteriológicos, antropométricos y de fltomejoramiento (Calinskiy Harabasz, 1974). Mientras que, para datos de percepción, se registra el uso del estadístico de Silueta (Kaufmany Rousseeuw, 1990). En datos relacionados a la genómica, como microarrays de ADN, la estadística de brechas ha sido utilizada por Tibshirani et al., 2001, el método de remuestreo Clest, para datos de expresión génica en estudios sobre cáncer (Dudoity Fridlyand, 2002) y, el índice de Conectividad, en datos de redes reguladoras de genes (Handly Knowles, 2005). En este trabajo, se estudió la performance de cuatro índices de validación para la búsqueda de estructura genética poblacional, en datos caracterizados por una alta densidad de marcadores moleculares de ADN del tipo SNP (polimorfismo de un solo nucleótido).
El objetivo de este trabajo es evaluar el desempeño de cuatro índices de validación, para precisar el número de grupo que determina la estructura genética poblacional subyacente.
MATERIALES Y MÉTODOS
Generación de datos
Se simularon datos con estructura genética po-blacional conocida, para evaluar la capacidad de los índices de detectar la cantidad de grupos esperados bajo simulación. Para ello, se trabajó con el paquete xbreed de R (Esfandyariy Sorensen, 2019), desarrollado para generar datos genéticos a partir de una población histórica, con distintos parámetros genéticos que imiten, en su conjunto, un determinado cultivo. En este caso, la simulación imitó un genoma de maíz. Las bases de datos fueron simuladas para marcadores moleculares del tipo SNP, considerando individuos diploides, a partir de una población histórica con 10 cromosomas. Los parámetros genéticos permiten simular alelos (marcadores de ADN) que, debido a su cercanía física en un cromosoma, se presentan juntos, de manera más frecuente que lo esperado por azar, lo que da lugar a la estructura genética poblacional. Así, los parámetros genéticos que permitieron simular los distintos escenarios fueron: número de individuos de la población inicial, número de marcadores moleculares, número de generaciones, tasa de mutación y heredabilidad, en sentido estricto. Se simularon nueve escenarios con diferentes configuraciones de estructura genética poblacional. Cada escenario se replicó 100 veces, simulando en cada réplica n=1000 individuos y p=80000 marcadores moleculares de tipo SNP El paquete xbreed, además de simular datos genómicos de marcadores moleculares, también permitió simular el feno-típico de los individuos, asociado a la información genómica. Luego, se tomaron muestras al azar de individuos fundadores de la población histórica y, para distinguir las subpoblaciones, se simularon generaciones posteriores para cada subpoblación reciente, indicando distintas varianzas fenotípicas y seleccionando a los individuos que conforman cada subpoblación, según los extremos fenotípi-cos altos y bajos. Los extremos fenotípicos fueron seleccionados, en este trabajo, a partir del primer y tercer cuartil de los valores obtenidos en la última generación. Una vez identificados los conjuntos de datos, se estimó el estadístico F, como una medida de diferenciación genética. De esta manera, se considera qué valores de diferenciación genética de Fst=0,03 indican un nivel bajo de diferenciación, entre grupos de individuos de la misma especie (poca separación entre subpoblaciones). Un valor de Fst=0,05 fue considerado como un nivel medio de diferenciación genética y, por último, un valor de Fst=0,07, fue considera un nivel alto de diferenciación genética, siguiendo los valores propuestos por Latch et al.(2006). Los tres niveles de diferenciación genética permitieron identificar distintos grados de EGP. La combinación de los tres niveles de diferenciación genética y el número de subpoblaciones (k=2, k=3 y k=10) simuladas dieron como resultado nueve escenarios biológicos, sobre los cuales se evaluó el desempeño de los índices de validación interna (Tabla 1).
En cada base de datos, la información provista por el marcador se codificó: con 0 el homocigo-ta y con 1 el heterocigota. Se eliminaron aquellos marcadores con frecuencia alélica menor a 0,01 y aquellos con más del 30 % de datos faltantes. Para calcular matrices de distancias, se utilizó el complemento a uno del índice de similitud de Jaccard (1-J), para transformar el índice de similitud en distancia (DJ). Dicha función, está implementada en R, con la función vegdist del paquete vegan, con método jaccard. Por lo tanto, DJ=1-J, donde J es el índice de similitud de Jaccard, basado en el número de co+presencias de marcador molecular en ambos individuos. Entonces, la expresión del índice de similitud de Jaccard es J=a/(a+b+c), con a, b y c, representando los recuentos de células de una tabla de contingencia, al cruzar la información recopilada a través de ambos perfiles genómicos en los individuos i y j, a es el número de co-presencias de marcadores SNP y las cantidades b y c son el número de presentes en uno y ausentes en el otro sujeto.
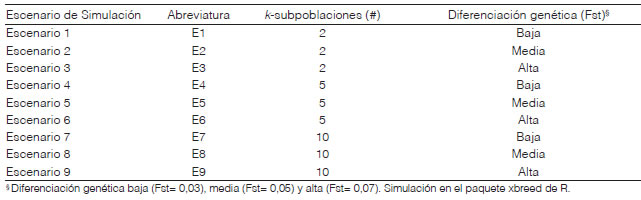
Tabla 1: Configuración de cada uno de los nueve escenarios de simulación, que surgen de tres tamaños de subpoblaciones (k) y tres niveles de diferenciación genética.
El ordenamiento de los individuos en el plano factorial, conformado por las dos primeras coorde-nadasde un Análisis de Coordenadas Principales, utilizando la distancia obtenida a partir del índice de Jaccard, permitió alcanzar la configuración de las subpoblaciones logradas para cada escenario de simulación. Esto permitió visualizar distintos niveles de separación (divergencia genética), lograda entre las subpoblaciones (Figura 1).
Índices de validación
Para validar el número óptimo de grupos en cada escenario de simulación, se utilizaron los índices CH (Calinski y Harabasz, 1974), Dunn (Dunn, 1974), Ancho de Silueta (Kaufman y Rous-seeuw,1990) y Conectividad (Handl y Knowles, 2005). Estos índices fueron calculados para cada simulación, a partir del agrupamiento obtenido por el método de agrupamiento bayesiano, que fue evaluado previamente por (Videla, 2021) como el método de menor tasa de error de clasificación para identificar la estructura genética poblacional subyacente en datos simulados y datos provenientes de ensayos agrícolas.
Esos índices son clasificados como índices de validación interna, es decir, sus algoritmos utilizan la información de los datos para determinar el número de grupos. El índice CH se basa en la dispersión entre las observaciones que fueron clasificadas en un mismo grupo, con relación a la distancia entre grupos. Esta relación es calculada para distintos números de posibles grupos. Luego, el valor más alto de la relación entre grupos respecto a la distancia dentro de grupo, indica el número de subpoblaciones consideradas como óptimas por el índice CH. El índice de Dunn combina la compactación (homogeneidad dentro del conglomerado) con el grado de separación entre conglomerados (Brock et al., 2008). Mayor valor de Dunn implica mayor varianza entre conglomerados y menor varianza dentro del conglomerado. El Ancho de Silueta mide la confianza con la que una observación es asignada a un grupo. Si ha sido bien asignada, tendrá valores cercanos a 1. La Conectividad está relacionada a la distancia entre observaciones vecinas en un mismo conglomerado: mientras menor valor de Conectividad, mejor es la clasificación. A continuación, se describe el algoritmo de cada uno de los índices de validación para comprender sus diferencias y su comportamiento, al seleccionar un número óptimo de grupos, a partir de la clasificación.
Índice CH
El índice de validación del número de grupos propuesto por Calinski y Harabsz (1974) fue denominado CH, y es una medida comparativa entre:la dispersión de grupos y la desviación dentro del agrupamiento, teniendo en cuenta la compactación promedio dentro de grupo. Dicha compactación, es lograda por tener individuos más parecidos entre sí dentro de cada grupo. La Ecuación 1 representa, matemáticamente, este algoritmo de validación, donde BG es la matriz de dispersión entre grupos; y la traza representa la suma de los elementos de la diagonal de dicha matriz, indicando la varianza entre grupos estimada, como la distancia de la media del grupo a la media general de las distancias entre todos los individuos (Ecuación 1).

Figura 1: Gráfico de dispersión de las dos primeras coordenadas principales, obtenidas a partir del análisis de coordenadas principales, con la distancia obtenida a partir del complemento a uno del índicede similitud de Jaccard, sobre una de las repeticiones de cada una de las bases simuladas. Cada base de datos simulada contiene 1000 individuos genotipados con 80K SNPs. Cada configuración representa uno de los nueve escenarios de simulación, que difieren en el número de k grupos: k = 2 (izquierda), k = 5 (centro) y k = 10 (derecha); y en la diferenciación genética: baja (arriba), media (centro) y alta (abajo). Cada individuo está representado por un punto. Los individuos que pertenecen al mismo grupo están representados con el mismo color.

donde k, es el número de grupos formado, n la cantidad de individuos dentro de cada grupo y |j la media del grupo. WG es la matriz de dispersión dentro de los grupos para los datos agrupados en k, grupos cuyos coeficientes son wjk|, donde i y j son individuos del mismo grupo k, y son calculados como el producto de la diferencia entre el valor observado del individuo i, en el k-ésimo grupo (V¡k|), y la media de su grupo con la diferencia del valor del j-ésimo individuos, con su media en el mismo grupo (V|k|). Así, w|k|, queda conformado por w|k|= (V¡k| - u¡k|)T (Vjk| - u|k|l),donde V¡k|,representa el vector fila i de la matriz de datos, y V|k|, el vector columna j, de la matriz de datos. Este producto representa una medida de correlación entre los individuos i y j, que se encuentran en el mismo grupo.
Ancho de Silueta
El Ancho de Silueta mide la confianza con la que una observación es asignada a un grupo, en función de la disimilaridad de dicho individuo, con respecto a los individuos asignados al mismo conglomerado. Luego, se compara dicha disimilari-dad, con las disimilaridades encontradas respecto al resto de los individuos en otro grupo. Se espera que, si los genotipos tienen alta similitud con los individuos de su mismo grupo, entonces, el individuo ha sido bien asignado a dicho grupo. Así, el valor del índice se acerca a 1, cuando más alta es la confianza de que el individuo haya sido bien asignado a ese grupo. La expresión de este índice se presenta en la Ecuación 6.
Dunn
El índice de Dunn es el cociente de la mínima distancia entre dos observaciones que no pertenecen a un mismo conglomerado (mindkk.), y la máxima distancia entre dos observaciones de un mismo conglomerado (maxDk ). La expresión de este
índice puede formularse de la siguiente manera:


Conectividad
El índice de Conectividad alcanza su valor óptimo cuando el valor es el más pequeño que obtuvo al recorrer distinto número de grupos. En ese umbral del valor mínimo obtenido,se determina el número óptimo de grupos a formar. Para estimar el valor de conectividad, este índice construye una matriz de vecinos, donde coloca un cero si son vecinos y, otro valor, si no lo son. Por ello, mientras más pequeño es su valor, mayor es la conectivi-dad. Así, la Ecuación 7 presenta la fórmula para su estimación, donde si nn es el -ésimo vecino más
![]()
Comparación de los índices de validación
En este trabajo, el cálculo de los índices de validaciónse realizó a partir de vectores de asignación, obtenidos con el Método Bayesiano (MB), implementadoen el paquete LEA, en R (Frichoty Frangois, 2015), para cada réplica de cada escenario, variando la cantidad de grupos, al calcular el índice desde k=2 hasta k=15. En este Método Bayesiano de agrupamiento difuso, propuesto por Pritchard et al. (2000), implementado en el software SRUCTURE, los genotipos se asignan de manera probabilística a un grupo, basado en las cadenas de Markov. Dado que el Método Bayesiano estima una probabilidad de pertenencia de un individuo a uno de los k grupos configurados, cuanto más alta sea la similitud de un individuo con un grupo, más alta será la probabilidad asignada. Puede suceder que el individuo sea asignado a dos o más conglomerados, simultáneamente, si el genotipo indica una mezcla de patrones moleculares; por ello, es denominado agrupamiento difuso. En este método, como en otros bayesianos de conglomeración difusa, las probabilidades a posteriori indican la incertidumbre de la asignación de conglomerados. Cabe destacar que, este método, logró una perfecta clasificación de los individuos en todos los escenarios, cuando el k indicado fue el mismo que el simulado (k=2 para E1, E2 y E3; k=5 para E4, E5 y E6; k=10 para E7, E8 y E9).
Como criterio de comparación de los índices implementados, se calculó su tasa de error de clasificación. Para conocer la precisión en el número de grupos sugeridos por el índice de validación, se varió el número de grupos k, es decir, se forzó a realizar agrupamientos diferentes a la configuración simulada. Luego, se evaluó, en cada réplica y para cada escenario, el número asignado según el índice y el número esperado según el verdadero número de grupos. Es importante recordar que el número correcto de grupos subyacente en un conjunto de datos, no suele ser conocido a priori. Dicho de otra manera, es usual que los individuos que conforman el conjunto de datos reales no tengan una clasificación previa. A través de la simulación realizada en este trabajo, se consideró como verdadero al número de grupos generados bajo la configuración de la simulación. De esta manera, se verificó la variación resultante de la precisión de los índices de validación, para sugerir el número óptimo de grupos. La diferencia entre el número de grupos simulados y el número de grupos sugerido por el índice fue considerada una tasa de error de clasificación, que denominamos Error de tipo III (E III). Además, se identificó el error cometido por estimar un número de grupos superior al simulado (E III+) o, por el contrario, subestimar el número de grupos, al sugerir una cantidad inferior al esperado bajo simulación (E III-). A partir del error tipo III (E III), discriminando entre la sobreestimación del número de grupos (E III+) y la subestimación (E III-), se comparó la capacidad de cada índice, para sugerir el número correcto de grupos subyacentes.
RESULTADOS Y DISCUSIÓN
Para los escenarios de simulación con dos subpoblaciones (E1, E2 y E3), los cuatro índices de selección del error de tipo III de sobreestimación (E III+) del número de grupos fue nulo (0 %). Es decir, todos los índices sugirieron que k era igual a 2, para las 100 réplicas de cada escenario, cuya configuración simulada era de k igual a 2. En este caso, no fue posible estimar E III-, ya que los índices parten del supuesto de que, al menos, hay dos grupos. De allí, la importancia del resultado hallado en los escenarios E1, E2 y E3, donde ningún índice sugirió mayor cantidad de grupos que el previsto por simulación. En los escenarios de simulación, que incluyeron cinco subpoblaciones (k=5, escenarios E4, E5 y E6), los índices de validación del número seleccionado de grupos tuvieron tasa de sobrestimación (EIII+) nula con Silhouette y Dunn. Es decir, ambos índices indicaron el número correcto de subpoblaciones (k=5), en las 100 réplicas de cada escenario. Respecto al índice CH, el número verdadero de grupos fue identificado en un promedio del 91 % de las réplicas, con un intervalo entre 86 % y 97 %, según el escenario. Es decir, en la mayoría de las réplicas, no hubo error al sugerir el número de grupos correctos. Contrario a lo observado con los otros índices, en el caso del índice de Conectividad, un promedio del 95 % de las veces, subestimó el número de grupos [94-96 %], sugiriendo dos subpoblaciones como número óptimo de grupos en la mitad de los casos.
Dado que, en el 52 % y 55 % de las réplicas, no sólo subestimó el número de grupos, sino que indicó dos grupos en lugar de cinco. En los escenarios de simulación con diez subpoblaciones (k=10, E7, E8 y E9), los índices de Silhouette y Dunn tuvieron errores de tipo III de sobreestimación, y de subestimación nulos. Mientras que CH, subestimó (EIII-) el número de grupos entre un 12 % a un 17 %, y el índice de conectividad cometió Error de tipo III- en el 100 % de las veces (Tabla 2).
A pesar de que, para los escenarios con k=2, el índice de Conectividad tuvo tasa de error nula, debiéramos considerar la posibilidad de que el índice de validación denominado Conectividad, no sea suficientemente robusto para estimar el número óptimo de grupo cuando existen más de dos subpoblaciones dado a que, este índice, considera la distancia entre observaciones de un mismo grupo. Es decir que, grupos muy compactos, tenderán a disminuir la Conectividad, sin importar la separación entre ellos. Dicho de otra manera, el índice de Conectividad estima su valor a partir de la distancia de cada individuo con su y-ésimo vecino más cercano. Si el vecino pertenece a su grupo, entonces, asigna un cero a la suma de valores que conforman el índice (Ecuación 7), por el contrario, si el vecino pertenece a otro grupo, suma 1/j. Así, mientras más individuos cercanos -a menor distancia- pertenezcan a un mismo grupo, más pequeño será el valor de la Conectividad. De allí que, su criterio de optimización es que, el valor más pequeño de Conectividad sugiere el número de grupos. Así, cuanto menor sea la cantidad de grupos conformados por un método de agrupamiento, menor será el valor de Conectividad y, por ello, tenderá a sugerir un número menor de grupos que el verdadero. En nuestro estudio de simulación, al configurar cinco grupos, el índice indica dos, la mayoría de las veces, con los distintos métodos de agrupamiento. Esto puede explicarse porque asigna más cantidad de valores distintos de cero, debido al aumento de grupos, es decir, de individuos no vecinos que sumaran 1/y, y no cero. Así, para un conjunto de datos de igual cantidad de individuos, mientras mayor sea el número de grupos, más grande será el valor del índice de Conectividad, por la manera en que el mismo se construye (Ecuación 7). Los índices Dunn y Silueta indicaron correctamente el número óptimo de grupos en las 100 réplicas de todos los escenarios de simulación. Mientras que el índice CH y Conectividad solo con los escenarios de dos subpoblaciones (E1, E2 y E3). El índice CH seleccionó correctamente el número de grupos, más del 80 % de las veces en el que fue aplicado en los escenarios con cinco y diez subpoblaciones (E4-E9). Mientras que Conectividad tuvo una eficiencia menor al 10 %, es decir, solamente en un 10 % de las veces indicó el número correcto de grupos esperado bajo la simulación. En estos últimos escenarios de simulación con k=5 y 10 subpoblaciones, el índice CH seleccionó mayor cantidad de veces el número de grupos adecuado, mientras fuera menor el nivel de diferenciación genética (Figura 2). En el estudio de simulación de Latch et al. (2006), donde evaluaron el desempeño de los softwares STRUCTURE, BAPS y PARTITION, para identificar la subestructura de la población, los autores concluyeron que para niveles de diferenciación genética iguales o superiores a Fst=0,03, STRUCTURE estimó correctamente el número de grupos utilizando el índice Ak propuesto por Evanno et al. (2005). Teniendo en cuenta que nuestros escenarios de simulación tienen divergencia genética igual o superior a Fst=0,03, los resultados de los índices Dunn y Silueta hallados, en este trabajo, coinciden con los obtenidos por los autores del trabajo mencionado.
La identificación del número subyacente de grupos existentes en una población ha sido una pregunta de investigación desde los comienzos de la estadística, con el objetivo de dar respuesta a genetistas, respecto de la estructura que se forma por similitudes entre individuos de una o más poblaciones. A lo largo de la historia, se han propuesto numerosos algoritmos de clasificación que contemplaban tanto la naturaleza de los datos, como la capacidad de cálculo computacional disponible. Así, hemos pasado de métodos jerárquicos que podían ser aplicados sobre una amplia disposición de métricas de distancias, las cuales podían seleccionarse según la naturaleza de la variable con la que se trabajara. De este modo, con variables de naturaleza continua, la métrica más utilizada es la distancia euclídea por la propiedadde ultramétrica que le confiere robustez. Mientras que, si se trabaja con variables de naturaleza discreta, puede aplicarse el coeficiente de Pearson y la distancia de Excoffier (Excoffier et al., 1992), derivada del índice de similitud emparejamiento simple o simple matching, cuando la naturaleza de la variable es binaria. En el caso particular de datos binarios, se ha propuesto un amplio rango de índices de similitud, que permiten estimar el parecido entre los individuos, según se ponderen las co-presencias simultaneas, las co-ausencias simultaneas de manera conjunta o independiente (Bruno et al., 2003). Luego, los índices de similitud pueden ser transformados mediante funciones a distancia, sin pérdida de generalidad. Para situaciones donde existen mezclas del tipo de variables, la propuesta por Gower (1971) ha sido, hasta el momento, la única que admite este tipo de información combinada de variables continuas y discretas. Estas métricas han demostrado ser potentes para la estimación de la distancia entre individuos, y generar agrupamien-tos con diferentes algoritmos de clasificación jerárquicos, en un contexto de dimensión más reducido respecto al evaluado en este trabajo.

Tabla 2: Tasa de error de tipo III de subestimación (E III") y tasa de error de tipo III de sobreestimación (E III+) del número de grupos para cuatro índices de selección para una estructura genética poblacional simulada con k=2, 5 y 10 subpoblaciones: nivel bajo, medio y alto de diferenciación genética
Figura 2: Gráficos de dispersión de la clasificación correcta (cantidad de réplicas en las que el índice seleccionó correctamente el número de grupos) de cuatro índices de validación: CH, Conectividad, Dunn y Silueta, en nueve escenarios de simulación con 100 réplicas cada uno
CONCLUSIONES
Con el advenimiento de tecnologías capaces de generar bases de datos de mayor dimensión, tanto por el aumento en el número de variables por medir como en el número de individuos, los métodos no jerárquicos surgieron como técnicas más eficientes en el tiempo de cálculo computacional. En particular, en áreas donde el interés radicaba en la delimitación de zonas de manejo homogénea, utilizando datos de sensores remotos (Córdoba et al., 2019). Es decir, desde los comienzos del análisis de datos, los métodos de agrupamiento o clasificación han sido aplicados en diversas áreas (medicina, agronomía, ecología, genética, son algunas entre las ciencias vivas), como una herramienta objetiva para comprender el ordenamiento de los datos. Sin embargo, pocas veces se han llevado adelante estudios de comparación de la performance de los métodos en contextos particulares de datos, como en este caso de información proveniente del genoma de individuos. Actualmente, la disponibilidad de datos genómicos de 80K, como los simulados en este trabajo, es cada vez más frecuente. En escenarios con un número de grupo distinto de k = 2, el índice de conectividad tuvo el error de subestimación más alto del número de grupos, independientemente del método utilizado. Nuestros hallazgos sugieren que, en un contexto de alta cantidad de grupos subyacentes, los índices de Dunn y Silhouette tienen mejor desempeño para identificar el verdadero número de grupos subyacentes, cuando la clasificación de los individuos fue realizada a partir del método ba-yesiano. Este hallazgo ofrece una alternativa sólida para revelar la EGP existente, facilitando, de esta manera, los estudios de población y las estrategias de mejoramiento de cultivos.
BIBLIOGRAFÍA
Balzarini, M., Teich, I., Bruno, C. y Peña, A. (2011). Making genetic biodiversity measurable: A review of statistical multivariate methods to study variability at gene level. Revista de la Facultad de Ciencias Agrarias, 43(1), 261-275. http://www.scielo.org.ar/pdf/refca/v43n1/ v43n1a20.pdf
Brock, G., Pihur, V, Datta, S. y Datta, S. (2008). clValid: An R Package for Cluster Validation. Journal of Statistical Software, 25(4), 1-22. https://doi.org/10.18637/jss. v025.i04
Bruno, C., Balzarini, M. y Di Rienzo, J. (2003). Comparación de medidas de distancias entre perfiles RAPD. Journal of Basic & Applied Genetics, 15(1), 29-32. https:// www.researchgate.net/publication/283569265
Caliski, T., y Harabasz, J. (1974). A dendrite method for cluster analysis. Communications in Statistics-theory and Methods, 3(1), 1-27. https://doi. org/10.1080/03610927408827101 Charrad, M., Ghazzali, N., Boiteau, V. y Niknafs, A. (2014). NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. Journal of Statistical Software, 61(6), 1-36. https://doi.org/10.18637/jss. v061.i06
Córdoba, M., Paccioretti, P A., Giannini Kurina, F., Bruno, C. I. y Balzarini, M. G. (2019). Guía para el análisis de datos espaciales en agricultura. Serie Estadística Aplicada. http://hdl.handle.net/11336/128391 Dudoit, S. y Fridlyand, J. (2002). A prediction-based resampling method for estimating the number of clusters in a dataset. Genome biology, 3(7), 1-21. https://doi.org/10.1186/gb-2002-3-7-research0036 Dunn, J. C. (1974). Well-separated clusters and optimal fuzzy partitions. Journal of cybernetics, 4(1), 95-104. https://doi.org/10.1080/01969727408546059 Eick, C. F, Vaezian, B., Jiang, D. y Wang, J. (2006). Discovery of Interesting Regions in Spatial Data Sets
Using Supervised Clustering. En J. Fürnkranz, T. Scheffer, (Ed.), Knowledge Discovery in Databases: PKDD 2006. PKDD 2006. Lecture Notes in Computer Science (127-138). Springer, Berlin, Heidelberg. https://doi.org/10.1007/11871637_16 Esfandyari, H. y Sorensen, A. C. (2019). xbreed: An R package for genomic simulation of purebred and crossbred populations. https://cran.microsoft. com/snapshot/2020-04-05/web/packages/xbreed/ vignettes/xbreedvignette.pdf Evanno, G., Regnaut, S. y Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14(8), 2611-2620. https://doi.org/10.1111Zj.1365-294X.2005.02553.X
Excoffier, L., Smouse, P. E. y Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics, 131(2), 479-491. https://doi.org/10.3354/meps198283 Frichot, E. y Frangois, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6(8), 925-929. https://doi.org/10.1111/2041-210X.12382 Gower, J. C. (1971). A general coefficient of similarity and some of its properties. Biometrics, 27(4), 857-871. https://doi.org/10.2307/2528823
Halkidi, M. y lordanis, K. (2011). Online clustering of distributed streaming data using belief propagation techniques. En 2011 IEEE 12th International Conference on Mobile Data Management, 216-225. Lulea, Sweden. https://doi.org/10.1109/MDM.2011.63 Halkidi, M., Vazirgiannis, M. y Batistakis, Y. (2000). Quality scheme assessment in the clustering process. En D. A. Zighed, J. Komorowskiy, J. Zytkow (Eds.), Principles of Data Mining and Knowledge Discovery. PKDD 2000. Lecture Notes in Computer Science,1910 (265-276). https://doi.org/10.1007/3-540-45372-5_26 Handl, J. y Knowles, J. (2005). Exploiting the Trade-off The Benefits of Multiple Objectives in Data Clustering. En C. A. CoelloCoello, A. H. Aguirre y E. Zitzler (Eds.), Evolutionary Multi-Criterion Optimization. EMO 2005. Lecture Notes in Computer Science, 3410 (547-560). Springer. https://doi.org/10.1007/978-3-540-31880-4_38
Hartigan, J. A. (1975). Clustering Algorithms. John Wiley & Sons, Inc.
Jombart, T., Devillard, S. y Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC genetics, 11 (1), 1-15. https://doi.org/10.1186/1471-2156-11-94
Kaufman, L. y Rousseeuw, P. J. (1990). Finding Groups in Data: An Introduction to Cluster Analysis. John Wiley & Sons, Inc.
Latch, E. K., Dharmarajan, G., Glaubitz, J. C. y Rhodes, O. E. (2006). Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conservation Genetics, 7(2), 295-302. https://doi.org/10.1007/s10592-005-9098-1
Lawson, D. J., van Dorp, L. y Falush, D. (2018). A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nature Communications, 9(1), 1-11. https://doi.org/10.1038/s41467-018-05257-7
Lee, E. A. y Tracy, W. F. (2009). Modern maize breeding. Handbook of Maize, 141-160. https://doi. org/10.1007/978-0-387-77863-1_7
Odong, T. L., van Heerwaarden, J., Jansen, J., van Hintum, T. J. L. y Van Eeuwijk, F. A. (2011). Determination of genetic structure of germplasm collections: Are traditional hierarchical clustering methods appropriate for molecular marker data? Theoretical and Applied Genetics, 123(2), 195-205. https://doi.org/10.1007/ s00122-011-1576-x
Peña-Malavera, A., Bruno, C., Fernandez, E. y Balzarini, M. (2014). Comparison of algorithms to infer genetic population structure from unlinked molecular markers. Statistical Applications in Genetics and Molecular Biology, 13(4), 391-402. https://doi.org/10.1515/sagmb-2013-0006
Pritchard, J., Stephens, M. y Donnelly, P. (2000). Inference of population structure using multil ocusgenotype data. Genetics, 155(2), 945-959. https://doi.org/10.1093/ genetics/155.2.945
Rezaee, M. R., Lelieveldt, B. P. y Reiber, J. H. C. (1998). A new cluster validity index for the fuzzy c-mean. Pattern Recognition Letters, 19(3-4), 237-246. https://doi.org/10.1016/S0167-8655(97)00168-2
Salvador, S. y Chan, P. (2004). Determining the number of clusters/segments in hierarchical clustering/ segmentation algorithms. En 16th IEEE international conference on tools with artificial intelligence. IEEE, 576-584. Copenhagen, Denmark. https://doi. org/10.1109/ICTAI.2004.50
Tibshirani, R., Walther, G. y Hastie, T. (2001). Estimating the number of clusters in a data set via the gap statistic. Journal of the Royal Statistical Society: Series B (Statistical Methodology), 63(2), 411-423. https:// doi.org/10.1111/1467-9868.00293
Videla, M. E. (2021). Evaluación de algoritmos de agrupamientos para inferir estructura genética poblacional en datos genómicos. Tesis de Maestría publicada. Universidad Nacional de Córdoba, Córdoba, Argentina. http ://hd l. hand le. net/11086/20184

