











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
Desde el punto de vista terapéutico las hembras preñadas, al igual que los gerontes y neonatos, son consideradas una población especial. Su terapéutica es una preocupación y un desafío para los profesionales médicos, tanto veterinarios como humanos.
Lo que transforma en especiales a estas poblaciones son las características fisiológicas y bioquímicas propias que conducen a cambios en la farmacocinética de los fármacos administrados que pueden afectar de manera significativa su eficacia terapéutica.
Es importante remarcar que la bibliografía sobre este tema en veterinaria es muy escasa, especialmente en lo referente a los cambios en la farmacocinética de los fármacos administrados a la hembra preñada. Si bien existen reportes sobre los efectos clínicos de fármacos administrados durante la preñez se refieren a casos individuales y se enfocan exclusivamente a la evolución de la patología involucrada. Esta situación nos obliga a recurrir a bibliografía de medicina humana, la cual no siempre es extrapolable a las situaciones clínicas observadas en la clínica veterinaria.
La farmacodinamia es la rama de la farmacología que estudia los cambios que los fármacos son capaces de provocar en los organismos vivos. Si bien estos pueden ser extensos, son cambios fundamentalmente cuantitativos (estimular o inhibir una función biológica) y en ningún caso cualitativos (no existe ningún compuesto que induzca a una célula a realizar una función que no le es propia).
La farmacocinética estudia los cambios que el organismo induce en los fármacos. Permite estudiar y cuantificar el tránsito de las moléculas a través del organismo y engloba básicamente 4 procesos: absorción, distribución, metabolismo y excreción (ADME).
A efectos de cuantificar los procesos farmacocinéticos se generan los llamados parámetros cinéticos que reflejan la extensión o cuantía de cada uno de los procesos durante la disposición plasmática de fármacos.
Los parámetros mas descriptivos y utilizados para los cálculos de dosis o para la construcción de los regímenes de dosificación son:
Biodisponibilidad (F). Es el porcentaje de la dosis que accede a la circulación sistémica tras la administración extravascular. Se asume que las administraciones intravasculares tienen una biodisponibilidad del 100%.
Área bajo la curva concentración plasmática de fármaco vs tiempo (AUC).Refleja la exposición sistémica de fármaco y permite evaluar la biodisponibilidad y la eficacia.
Volumen de distribución (Vd). Refleja la capacidad de los fármacos de distribuirse a través del organismo; su valor determina la concentración máxima en sangre (Cmax).
Aclaramiento (Clearance (CLB). Refleja la capacidad del organismo de deshacerse de los fármacos. Este parámetro no diferencia metabolismo y excreción; Está determinado por la capacidad de metabolizar fármacos (fundamentalmente del hígado) así como de la capacidad de excretar del riñón. Es uno de los más importantes para calcular la dosis.
Vida media de eliminación. Es un parámetro híbrido (refleja distribución y eliminación) generalmente sobrestimado tiene utilidad para calcular los intervalos interdosis.
Es importante recordar que para cumplir el objetivo terapéutico planteado al seleccionar un tratamiento es necesario diseñar un régimen de dosificación. De acuerdo al objetivo propuesto se podrá requerir un tratamiento monodosis o multidosis.
En ambos casos lo que permite predecir la eficacia es la concentración sanguínea del fármaco que debe ubicarse en el interior de la llamada ventana terapéutica. Esta ventana está delimitada por la concentración mínima efectiva y la máxima tolerada. El tiempo de permanencia en ventana refleja la duración del efecto y determinará la selección del intervalo interdosis (Figura 1).
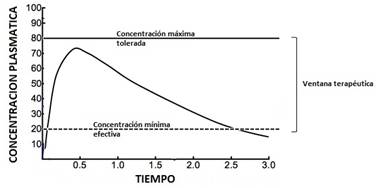
Figura 1 Curva de disposición plasmática de un fármaco tras su administración monodosis por vía extravascular.
En el caso de la administración de dosis repetidas (multidosis) quien permite predecir el efecto terapéutico es la llamada concentración en estado estacionario (Css) (Figura 2).
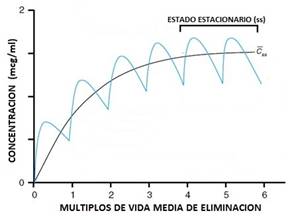
Figura 2 Curva de disposición plasmática de un fármaco tras su administración multidosis por vía extravascular.
El tiempo requerido para llegar a la concentración en estado estacionario está directamente relacionado con la vida media del fármaco, requiriéndose 5 vidas medias para alcanzarlo. Si la vida media del fármaco es larga, por ejemplo 12 horas, se necesitaran 60 horas (12 x 5) para lograr que la concentración plasmática se ubique en el interior de la ventana terapéutica. Para acortar este tiempo, que dejaría al paciente sin la medicación adecuada, se hace uso de la llamada dosis de ataque, que permite alcanzar tras la primera dosis la ventana terapéutica. Una vez logrado ese objetivo el tratamiento se continúa aplicando la dosis de mantenimiento.
Al observar las Figuras, notamos que uno de los parámetros más importantes es la concentración máxima (Cmax) que está determinada por la dosis administrada y el volumen de distribución del fármaco. La capacidad de un fármaco de distribuirse en mayor o menor grado es reflejo de su capacidad de atravesar membranas; por lo tanto las moléculas liposolubles no ionizadas se distribuirán en mayor grado. Por otro lado, como los fármacos cuando llegan a la circulación pueden unirse a las proteínas plasmáticas (principalmente albúminas) por lo que existirán 2 fracciones, la libre y la conjugada a proteínas; dado que las proteínas por su tamaño no pueden atravesar las membranas normales (no inflamadas), solo la fracción libre saldrá del torrente sanguíneo siendo considerada desde el punto de vista farmacológico la fracción activa.
La velocidad de disminución de las concentraciones en sangre refleja la capacidad del organismo de eliminar el fármaco. Los fármacos son eliminados del organismo por diferentes procesos. Los hidrosolubles, se eliminan fundamentalmente por filtración y/o secreción activa a nivel renal, mientras que los liposolubles requieren metabolismo que los transforma en compuestos (metabolitos) hidrosolubles/polares. El metabolismo se lleva a cabo en muchos órganos y tejidos, sin embargo, el hígado es el más importante. Entre las diferentes reacciones metabólicas de fármacos la oxidación, a través de la familia de citocromos P450, es la más común.
La hembra preñada
Es importante remarcar que al referirse a la terapéutica de la hembra preñada en realidad nos referimos a la unidad materno-fetal, ya que todo fármaco que se administre a la hembra preñada va a provocar efectos (mayores o menores, nocivos o inocuos) en el embrión/feto (Cooper et al. 1999).
La terapéutica de la hembra preñada es, como se mencionó previamente, un desafío. Ante la llegada de una hembra preñada al consultorio, hay ciertas preguntas que el clínico debe hacerse; la primera: si es imprescindible tratarla farmacológicamente y la segunda, si la patología permite (aplicando medidas de contención) retrasar el inicio del tratamiento hasta finalizada la preñez.
La hembra preñada, desde el punto de vista farmacocinético, es una paciente con importantes cambios fisiológicos que modificarán de manera significativa el perfil de disposición de los fármacos (Pavek et al. 2009).
Cambios fisiológicos durante la preñez con efecto sobre la farmacocinética de los fármacos
Los cambios fisiológicos más importantes (por sus consecuencias en los procesos farmacocinéticos) se observan a nivel cardiovascular (Pacheco et al. 2013). Éstos tienen un ritmo temporal, siendo máximos al día 40 de la preñez; posteriormente comienzan a descender hasta alcanzar los valores de la hembra no preñada el día 20-30 postparto (Ward et al. 2020).
El cambio más notorio es el aumento entre el 30 y el 40% del gasto cardiaco. Los mecanismos de este aumento no son claros; por un lado existe evidencia de una disminución de la resistencia vascular periférica inducida por las hormonas gestacionales que activa el sistema renina-angiotensina-aldosterona, conduciendo al incremento en la reabsorción de sodio a nivel renal y por ende, al aumento del volumen sanguíneo (Pacheco et al. 2013, Costantine 2014, Almeida et al. 2017).
Asimismo, se observa un cambio en los flujos sanguíneos regionales a los diferentes órganos, con un aumento a nivel genitourinario y piel y una disminución compensatoria en el músculo esquelético (Metcalfe et al. 1955).
El aumento del flujo sanguíneo de la piel tiene el objetivo de disipar el calor producido por el feto. El flujo sanguíneo hepático no sufre modificaciones a diferencia del renal que sufre un aumento importante, reflejado por el incremento de la tasa de filtración glomerular (Ginsburg y Duncan 1967).
Otro cambio fisiológico importante, relacionado al aumento del volumen sanguíneo, es el cambio en la distribución del agua corporal, con una expansión del espacio extracelular y, en menor medida, del intracelular (Pavek et al. 2009).
A nivel respiratorio, se observa una estimulación del centro respiratorio (inducida por progesterona) que conduce al aumento en la presión parcial de oxígeno con una disminución en la presión parcial de CO2 (para facilitar el pasaje del CO2 del feto a la madre) (LoMauro y Aliverti 2015). Para compensar, aumenta la excreción renal de bicarbonato, lo que conduce a una disminución del bicarbonato sérico con un leve aumento del pH sanguíneo; es decir que hay un estado de alcalosis respiratoria parcialmente compensada (Pacheco et al. 2013). Es importante remarcar que los estados de alcalosis pueden afectar la unión de los fármacos a las proteínas plasmáticas (Tillement et al. 1978).
Con respecto a la concentración plasmática de albúminas hay caída neta por cambios en el ciclo de síntesis/catabolismo hepático (Papich y Davis 1986).
Es importante remarcar, que no todos estos cambios tendrán consecuencias clínicamente apreciables (Loebstein et al. 1997). Se debe poner especial atención en los fármacos con ventana terapéutica estrecha, los cuales verán modificado su efecto clínico (aumentado o disminuido) lo que en muchos casos obligará a aplicar correcciones en el régimen de dosificación (Pavek et al. 2009).
Absorción de fármacos en la hembra preñada
Vías enterales. A nivel gastrointestinal se observan numerosos cambios fisiológicos con potencial capacidad de modificar la absorción de fármacos
Por acción de progesterona se induce la disminución de la velocidad de vaciado gástrico (Alqudah et al. 2022) y de la motilidad del intestino delgado (Lawson et al. 1985). Estos cambios pueden provocar un aumento del tiempo de aparición de la concentración máxima (Tmax), lo que se va a traducir en un aumento del tiempo de latencia del efecto (Pacheco et al. 2013).
A nivel gástrico disminuye la secreción de hidrogeniones lo que conduce a una elevación del pH acompañado por un aumento en la producción de moco (Pavek et al. 2009). La consecuencia del cambio de pH va a afectar a los fármacos ácidos, que se van a absorber menos (porque van a estar más ionizados y por ende menos liposolubles), lo contrario pasará con los básicos (Rowland y Tozer 2011). Con respecto a los fármacos sensibles al pH ácido (como la eritromicina), la biodisponibilidad estará aumentada (Cooper et al. 1999).
Vías parenterales. En el caso de la administración intramuscular, por la diminución del flujo sanguíneo local, la absorción será lenta y escasa, lo que se reflejará en un Tmax más prolongado y una Cmax menor que la observada en la hembra no preñada (Krauer y Krauer 1977).
Lo contrario se observa a nivel subcutáneo, ya que el flujo sanguíneo a este nivel está aumentado, lo que conduce a una absorción mayor y más rápida reflejada por un Tmax más corto y una Cmax mayor (Soldin y Mattison 2009).
Distribución de fármacos en la hembra preñada. Los cambios en el proceso de distribución son consecuencia directa de los cambios a nivel cardiovascular y de la redistribución del agua corporal. Considerando el aumento del volumen plasmático, con el consecuente aumento del espacio vascular y extravascular, se creará un mayor espacio para la dilución de los fármacos. En consecuencia el volumen de distribución se verá aumentado (Pavek et al. 2009).
En el caso de los fármacos liposolubles con alta unión a proteínas plasmáticas, la baja concentración de albúminas incrementará aún más el volumen de distribución (Frederiksen 2001) incrementando el acceso del fármaco al feto (Rebuelto y Loza 2010).
En un esquema de administración monodosis, y considerando que la dosis administrada es la indicada para la hembra no preñada, el aumento del volumen de distribución conducirá a una disminución de la Cmax y del AUC, lo que resulta en un acortamiento del tiempo durante el cual las concentraciones plasmáticas estarán por encima de la concentración mínima efectiva (Figura 3).
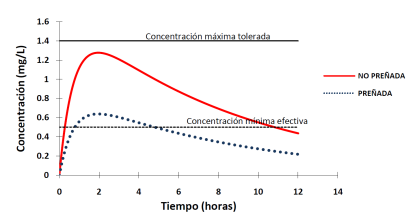
Figura 3 Consecuencias de las alteraciones del proceso de distribución sobre la curva de disposición plasmática de un fármaco tras su administración monodosis por vía extravascular.
En un esquema de administración multidosis, el más utilizado en la práctica, el aumento del volumen de distribución, conduce a la disminución de la concentración en estado estacionario (Figura 4).
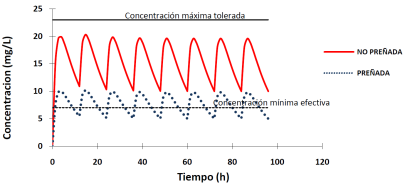
Figura 4 Consecuencias de las alteraciones del proceso de distribución sobre la curva de disposición plasmática de un fármaco tras su administración multidosis por vía extravascular.
Una solución para mantener las concentraciones plasmáticas por encima de la mínima efectiva sería el aumento de la dosis administrada. Sin embargo, la corrección no es tan simple ya que, como se mencionó previamente, se podría generar toxicidad en el feto (Abdelwahab et al. 2022). En la segunda parte de la revisión, se discutirán las posibles soluciones a estas situaciones para los fármacos más indicados en las hembras gestantes.
Eliminación de fármacos en la hembra preñada
Los fármacos se metabolizan con el objetivo de aumentar su excreción, por lo que este proceso conduce a un aumento de la hidrosolubilidad/polaridad de las moléculas. Los fármacos altamente hidrosolubles no sufren un metabolismo importante; por el contrario cuanto más liposoluble es un fármaco mayor será su metabolismo (Rowland y Tozer 2011).
Las reacciones metabólicas se dividen en dos categorías: de fase 1 y de fase 2. La oxidación es la más importante dentro de la fase 1 debido a que es la más común, es inducible y los metabolitos formados pueden resultar inactivos, menos activos o más activos que el fármaco madre. Dentro de las reacciones de fase 2, también llamadas de conjugación, la más importante es la glucuronoconjugación (Rowland y Tozer 2011).
El proceso de oxidación es mediado por diferentes enzimas. Las más importantes son las citocromo P450, una familia amplia de enzimas que se diferencian por sus sustratos.
Estas citocromo son modificadas de distinta manera y en distinto grado por los estrógenos y la progesterona (Noyola-Martínez et al. 2019). La mayoría son inducidas, como la 2D6, que corresponde en el perro a la 2D15, la 3A y la 2C9, que corresponde en el perro al 2C21/41 (Antonovic y Martinez 2011, Feghali et al. 2015). La inducción de estas enzimas conduce al aumento del aclaramiento (clearance) y por lo tanto, a la disminución de la vida media (Ryu y Hebert 2022). Algunos ejemplos de fármacos que se metabolizan por citocromos son fenobarbital, cloranfenicol, doxiciclina, dexametasona, clorpromacina, diazepam.
Si bien la mayoría de las citocromo son inducidas durante la preñez, una de ellas la 1A2 es inhibida; por lo tanto para los fármacos que son sustrato de esta vía metabólica, como naproxeno, paracetamol o las metilxantinas, se observará una disminución del aclaramiento, con el correspondiente alargamiento de la vida media (Noyola-Martínez et al. 2019, Pariente 2016).
Las consecuencias de la inducción enzimática en la eficacia de los fármacos son complejas y solo pueden predecirse si se conoce la actividad farmacológica de los metabolitos (Lynch y Neff 2007). Como se mencionó previamente, los metabolitos formados pueden tener mayor, menor o ninguna actividad farmacológica en relación a la droga madre, por lo tanto una inducción del metabolismo puede resultar en mayor (enrofloxacina) o menor actividad terapéutica (lincomicina) (Lynch y Neff 2007).
En la hembra preñada la excreción también estará profundamente modificada. Los estrógenos reducen el flujo biliar, entonces para aquellos fármacos que se excretan por bilis y sufren ciclo enterohepático habrá un retraso en la excreción, es el caso de las tetraciclinas (Anderson 2005).
A nivel renal, el importante aumento del flujo renal y de la filtración glomerular (que llega al 160% al final de la gestación) conducirá a un aumento neto de la excreción, en especial de los fármacos hidrosolubles (por ejemplo los aminoglucósidos) (Frederiksen 2001).
Los cambios en la eliminación de fármacos en la hembra preñada ya sea, por inducción /inhibición del metabolismo o incremento de la excreción conducirá a dos escenarios, el aumento y la disminución significativa del aclaramiento. Estos cambios, como se muestra a continuación conducen a la modificación del perfil plasmático de los fármacos (Figura 5 y 6).
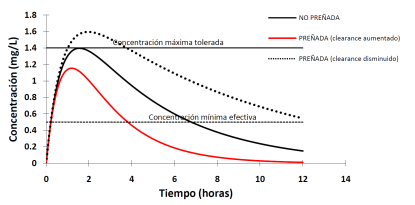
Figura 5 Consecuencias de las alteraciones del proceso de eliminación sobre la curva de disposición plasmática de un fármaco tras su administración monodosis por vía extravascular.
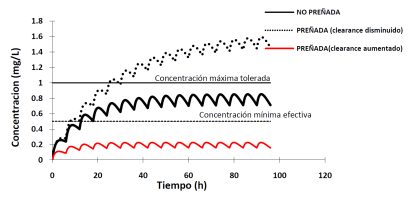
Figura 6 Consecuencias de las alteraciones del proceso de eliminación sobre la curva de disposición plasmática de un fármaco tras su administración multidosis por vía extravascular.
La inhibición del metabolismo se va a reflejar como un aumento de la vida media; las concentraciones plasmáticas estarán por encima de la mínima efectiva aproximadamente por el doble de tiempo que en la hembra vacía (Pavek et al. 2009). Es importante ser cuidadosos al extraer conclusiones sobre este cambio ya que, si el fármaco tiene metabolitos activos, el resultado sobre la eficacia es la disminución de la misma (Lynch y Neff 2007).
La inducción de los sistemas enzimáticos oxidativos o el incremento en la excreción renal (en caso de fármacos hidrosolubles que no sufren metabolismo) resulta en un acortamiento de la vida media. Sin embargo, si el fármaco en cuestión tiene metabolitos más activos que la droga madre el cambio se reflejará en un aumento de la eficacia (Feghali et al. 2015).
Corrección de los regímenes de dosificación
Existen dos estrategias para intentar reubicar el perfil de disposición plasmática del fármaco dentro de la ventana terapéutica: modificar la dosis o modificar el intervalo interdosis (Thummel et al. 2006).
En los diseños de dosificación monodosis, la reducción del tiempo de permanencia de las concentraciones de fármaco dentro de la ventana terapéutica, ya sea por el aumento del volumen de distribución o del aclaramiento podría ser corregido aumentando la dosis (Mehvar 1998). Sin embargo, no es aconsejable por el riesgo de aumentar la llegada de fármaco al feto.
En los esquemas multidosis la situación es más compleja, aunque debemos recordar que la corrección de dosis o intervalo solo es necesaria cuando la concentración plasmática escapa de manera significativa de la ventana terapéutica (Thummel et al. 2006).
Un aumento en la eliminación o en la distribución se refleja en concentraciones por debajo de la mínima efectiva. Mientras que, en el caso de una disminución en la eliminación las concentraciones superarán a la máxima tolerada (Mehvar 1998) (Figura 7).

Figura 7 Alteraciones de la curva de disposición plasmática de un fármaco tras su administración multidosis por vía extravascular que requieren corrección.
La selección del método de corrección dependerá del origen del cambio en el trazado. En el caso de una paciente con la eliminación aumentada es preferible reducir el intervalo interdosis, ya que como muestran las Figuras 8 y 9 la fluctuación de las concentraciones es menor comparado con el aumento de la dosis. Esto es consecuencia de que la fluctuación de las concentraciones depende de la relación intervalo interdosis/vida media (Mehvar 1998).
En pacientes con la eliminación disminuida por el contario, es aconsejable disminuir la dosis ya que la fluctuación será menor (Figuras 8 y 9).
Sin embargo, las generalizaciones no son siempre apropiadas, por esa razón en las siguientes secciones se discutirá la necesidad y el método de corrección más adecuado para cada fármaco en particular.
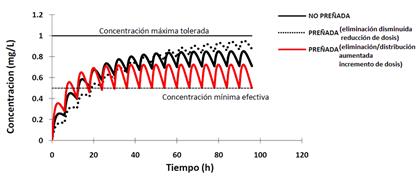
Figura 8 Modificación de la curva de disposición plasmática de un fármaco tras su administración multidosis por vía extravascular aplicando corrección de dosis.
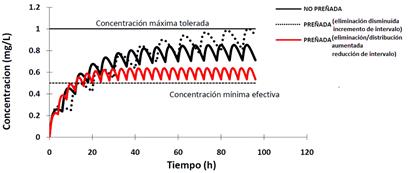
Figura 9 Modificación de la curva de disposición plasmática de un fármaco tras su administración multidosis por vía extravascular aplicando corrección de intervalo interdodis.
La unidad materno-fetal. La placenta, independientemente de su tipo (en el caso de la perra endoteliocorial), no se considera una barrera de amplia capacidad para el pasaje de fármacos al feto (Papich y Davis 1986, Audus 1999). Aún cuando posee enzimas metabolizadoras de fase 1 y 2 y transportadores del tipo P-glicoproteína y de aniones y cationes (que reforzarían su capacidad de barrera) con concentraciones circulantes de fármacos altas y/o sostenidas estos sistemas son rápidamente saturados (Rebuelto y Loza 2010).
Los fármacos liposolubles van a atravesarla fácilmente mientras que, los hidrosolubles, ionizados, polares o altamente unidos a proteínas plasmáticas lo van a hacer con mayor dificultad (Langston 2001). En general, lo único que limita el pasaje de fármaco desde la sangre de la madre al feto es el flujo sanguíneo y éste se incrementa de manera importante a medida que aumenta el tiempo de gestación (Ward 1989, Flaherty 2013).
Los fármacos que atraviesan la placenta van a llegar al feto a través de la sangre venosa umbilical; el 50% de la misma ingresa a la circulación hepática fetal y el resto atraviesa el ducto venoso (Dawes y Chowienczyk 2001). De esta manera, la mitad del fármaco podría ser metabolizado en el hígado fetal, ya que el feto a la semana 8 posee capacidad metabólica tanto de fase 1 y 2 (aunque más baja comparado con el feto humano) (Mathews 2008).
Tanto los metabolitos como los fármacos sin metabolizar difunden de regreso a la sangre materna. Sin embargo, como los metabolitos son hidrosolubles su eliminación por difusión a la sangre materna es significativamente más baja que la de los fármacos sin metabolizar (Langston 2001).
A partir del día 40 el riñón fetal tiene desarrollo suficiente para comenzar a excretar sustancias (incluidos metabolitos) al líquido amniótico (Langston 2001). Es importante remarcar que los metabolitos tanto los de fase 1 (desmetildiacepam, desmetiltramadol, etc) como los de fase 2 (morfina-6-glucurónido, tetraciclina glucurónido, etc) quedarán retenidos en el líquido amniótico lo que es una de las fuentes de acumulación de fármacos en los compartimientos fetales (Mathews 2008). Por otro lado, dado que el pH del feto es levemente más ácido que el de la madre, los fármacos liposolubles de naturaleza básica no van a difundir y también se acumularán en el feto (Mathews 2008).
Categorías terapéuticas utilizadas comúnmente en la hembra preñada. Es importante remarcar que solamente se debe iniciar un tratamiento cuando es indispensable. Como se mencionó previamente, en los casos que se pueda, se debe intentar evitar por todos los medios el tratamiento farmacológico sistémico.
La Food and Drug Administration (FDA) ha establecido, basado en estudios controlados en embarazadas y estudios en animales, cinco categorías (A, B, C, D, X) para indicar el nivel de riesgo de los fármacos de producir teratogenicidad (FDA 1979) (ver Tabla 1).Existen contados grupos de fármacos que podrían requerirse de manera inexorable aún durante la preñez, ellos son: los antiepilépticos, los analgésicos y los antimicrobianos.
Tabla 1 Clasificación de fármacos de acuerdo al riesgo de provocar efectos teratogénicos (FDA 1979).
| Categoría | Descripción | Ejemplos |
| A | Estudios controlados en animales y humanos no demuestran riesgo para el feto. | Acido ascórbico; Calcifediol; Calcio (excepto acetato), Calcitriol; Cianocobalamina; Colecalciferol ; Acido fólico; Folinato cálcico ; Gluconato de hierro ; Levotiroxina ; Piridoxina; Tirosina; Tocoferol |
| B | Estudios no controlados en animales no indican riesgo. No existen estudios en humanos. Se acepta su uso durante la preñez | Amoxicilina; Amoxicilina / Ácido Clavulánico; Ampicilina; Anfotericina; Azitromicina; Bencilpenicilina; Carnitina; Cefaclor; Cefadroxilo; Cefalexina; Cefalotina; Cefapirina; Cefazolina; Cefoxitina; Cefpodoxima; Ceftazidima ; Ceftriaxona; Cefuroxima; Cimetidina; Clindamicina; Clotrimazol; Cloxacilina; Eritromicina; Fosfomicina; Metronidazol; Nitrofurantoína; Penicilinas G y V; Permetrina; Sucralfato; |
| C | Estudios no controlados en animales demuestran efectos adversos en el feto. Su utilización debe analizarse valorando el costo beneficio | Acetazolamida; Albendazol; Amikacina; Bacitracina; Betametasona; Ciprofloxacina; Claritromicina; Clonazepam; Dexametasona; Espiramicina; Fentanilo; Furosemida; Gabapentina; Gentamicina; Griseofulvina; Itraconazol; Ketoconazol; Levofloxacina; Mebendazol; Meloxicam; Neomicina; Norfloxacina; Ofloxacina; Omeprazol; Rifampicina; Tramadol; |
| D | Estudios controlados demuestran evidencias de daño fetal. En ocasiones el beneficio puede superar el riesgo. Utilizar cuando no existe otra alternativa | Acido acetilsalicílico; Alprazolam; Bleomicina; Diazepam; Doxiciclina; Estreptomicina; Fenitoína; Fenobarbital ; Midazolam; Minociclina; Penicilamina; Tobramicina; Acido Valproico. |
| X | Estudios controlados demuestran evidencias de daño fetal. El riesgo potencial supera ampliamente el beneficio. Contraindicados durante la preñez. | Torvastatina; Estradiol; Estrógeno conjugado; Finasterida; Flurazepam; Fluvastatina; Isotretinoína; Lovastatina; Misoprostol; Nandrolona; Raloxifeno; Simvastatina |
Antiepilépticos. Si bien es un tema importante en medicina humana, en veterinaria su importancia es relativa. En primer lugar, porque ante una preñez en una hembra epiléptica la decisión más racional es el aborto. Sin embargo, existen situaciones en las cuales los tutores deciden continuar la preñez y en otros casos, hasta la buscan. Como sea, la obligación del médico veterinario es intentar llevar a término la preñez con el mínimo riesgo para la madre y la cría (en muchos casos, el veterinario se puede negar, pero los tutores la continúan sin apoyo médico, lo que en definitiva es más peligroso para la hembra y la cría).
Si bien existen numerosos fármacos antiepilépticos en el mercado, los más comúnmente utilizados (y estudiados) en caninos son la fenitoína, el fenobarbital y su pro-droga, la primidona. La complicación más importante del uso de antiepilépticos en la hembra preñada es que, excepto los bromuros, todos están clasificados como clase X, o sea con alto riesgo de provocar efectos deletéreos en el feto. Asimismo, y con la misma excepción, sufren metabolismo oxidativo hepático a través de las enzimas citocromo P450 las cuales, como se mencionó, son modificadas por los estrógenos y progesterona. Esta situación, sumada a la propia capacidad de estos fármacos de inducir el metabolismo hepático, genera situaciones clínicas difíciles de predecir, sobre las cuales los reportes son contradictorios (Dupont y Vercueil 2021).
Existen dos estrategias a seguir, la primera, cada vez más común en medicina humana, es reducir la dosis al mínimo posible y en algunos casos interrumpir el tratamiento hasta el parto (Barut 2021); la segunda es utilizar los bromuros, especialmente de potasio (más potente) (Beghi 2011). Los bromuros tienen la ventaja de no ser teratogénicos y no sufrir metabolismo hepático (HPRA 2019). Si bien se eliminan por riñón y su eliminación estaría aumentada (Baird-Heinz et al. 2012), los reportes empíricos sugieren que no es necesario modificar la dosis. Los bromuros tienen vida media muy larga (25 días), lo que implica que el tiempo necesario para alcanzar el estado estacionario es de alrededor de 4 meses, por lo tanto es imprescindible aplicar una dosis de ataque (Bhatti et al. 2015). Si bien hay distintos esquemas el más reportado consiste en la administración oral de 400 a 600 mg kg-1 día-1 divididos en 2 o tres administraciones, siempre con el alimento. Esta dosis se administra por alrededor de 5 días (se debe monitorear la concentración plasmática) y luego se continua con la dosis de mantenimiento que es de 20 a 30 mg kg-1 día-1 (Bhatti et al. 2015). Este esquema implica que los responsables de la mascota deberán planificar con antelación la preñez e iniciar el tratamiento al menos 1 mes antes del servicio.
Analgésicos. El dolor es otra situación clínica que puede presentarse en la hembra preñada y que debe ser tratada. En casos de dolor severo, la primera elección son los opioides (Mathews 2008). Dentro del grupo, los más estudiados en preñez en humanos son morfina, metadona e hidromorfona. Los reportes en caninos son escasos y se refieren al periodo periquirúrgico de las cesáreas.
Las dosis recomendadas durante la preñez son, para morfina 0,5-2 mg kg-1 IM, SC c 3-4 h, para metadona (está en el país una presentación para humanos, laboratorios LIF) 1-1,5 mg kg-1 IV, IM o SC c 4-6 h y para hidromorfona (Dolonovag, importado) 0,05-0,2 mg kg-1 IV, IM, SC c 2-6 h.
Otros opioides, como fentanilo, meperidina, butorfanol y nalbufina no están indicados; son más liposolubles y alcanzan mayores concentraciones en el feto (Cooper et al. 1999, WSAVA 2014).
La 2da opción, cuando es posible, son los anestésicos locales los más seguros y no teratogénicos (WSAVA 2014).
Los AINEs, los alfa2 agonistas y la ketamina están contraindicados en la hembra preñada. Los AINEs son teratogénicos e interfieren el desarrollo fetal (WSAVA 2014). Los alfa 2 agonistas reducen el flujo sanguíneo uterino y pueden provocar muerte embrionaria (Flaherty 2013) y la ketamina, aumenta el tono uterino (WSAVA 2014).
Antimicrobianos. De los numerosos grupos de los que se dispone para tratar procesos infecciosos en caninos, son pocos los indicados para el tratamiento de la hembra preñada.
Los antibióticos de primera línea en hembra preñada, son los betalactámicos (en especial, las penicilinas y las cefalosporinas de primera generación (cefalexina, cefradoxil, cefalotina y cefazolina) (Rebuelto y Loza 2010).
Los betalactámicos en general y las penicilinas en particular tienen un perfil cinético óptimo para uso en hembras preñadas (baja unión a proteínas plasmáticas y ausencia de metabolismo a nivel hepático). Asimismo, para el grupo no se reporta riesgo teratogénico (Mylonas 2011).
Las penicilinas se eliminan por secreción renal y si bien la eliminación renal de fármacos en las hembras preñadas está incrementada, no afecta en grado importante la eficacia (excepto en infecciones por bacterias de alto valor de CIM) (Heikkilä y Erkkola 1994). Las penicilinas, se clasifican como antimicrobianos con cinética de muerte bacteriana tiempo dependiente. Esto significa que la mayor eficacia se presenta cuando las concentraciones en sangre están por encima de CIM de la bacteria por al menos el 50% del intervalo interdosis. Es por esto, que solo en infecciones por bacterias con alto valor de CIM es necesario incrementar la dosis (Chow y Jewesson 1985, Einarson et al. 2001). De las penicilinas, las más estudiadas en hembras preñadas son la G y las bencilpenicilinas (amoxicilina y ampicilina). En los casos en que la bacteria productora de la infección ha sido identificada como Gram + no productora de betalactamasas, el compuesto de elección es la penicilina G (la más potente del grupo frente a bacterias Gram+) (Mylonas 2011). En casos de infecciones, como las de las vías urinarias, en las cuales se desconoce si la bacteria es Gram positiva o negativa, la elección es amoxicilina. Ampicilina en caninos tiene una absorción oral errática y reducida (Papich 2013).
Con relación a la administración de las asociaciones amoxicilina-ácido clavulánico o sublactam la evidencia empírica indica que el riesgo de provocar efectos teratogénicos es bajo (Papich y Davis 1986, Rebuelto y Loza 2010).
El grupo indicado para infecciones por bacterias Gram-negativas en hembras preñadas es el de los aminoglucósidos (excepto estreptomicina y neomicina).
Estos antimicrobianos tienen un perfil cinético similar al de los betalactámicos; son moléculas polares, con baja unión a proteínas plasmáticas, distribución exclusivamente al espacio extracelular, carecen de metabolismo hepático y se eliminan por filtración glomerular Desde el punto de vista farmacodinámico son del tipo concentración dependiente, esto significa que la mayor eficacia se presenta tras la administración de dosis altas una vez al día. Asimismo, esta estrategia de administración disminuye la incidencia de efectos tóxicos tanto a nivel del túbulo renal como en el oído interno.
Su administración en la hembra preñada debe ser cuidadosa, porque normalmente se requiere aumentar la dosis (la eficacia se asocia con una Cmax alta, y en la hembra preñada ésta se reduce) (Briggs et al. 2005). En preñeces avanzadas cuando el feto posee capacidad de filtración renal, el aminoglucósido se excretará al líquido amniótico y por su alta polaridad quedará retenido en el mismo con el peligro de acumulación y pasaje a todos los compartimientos fetales (Briggs 2014). De todos los compuestos, el de perfil más seguro es gentamicina (Papich y Davis 1986, Heikkila 1993).
Los macrólidos han sido utilizados en medicina humana por muchos años, sin embargo, debido a reportes contradictorios en referencia a su eficacia y seguridad, ya no se los considera de 1era elección (Fan et al. 2019, Fan et al. 2020). Los macrólidos son fármacos básicos y de gran tamaño por lo que su pasaje a través de la placenta es bajo; eritromicina es el compuesto que mayor pasaje reporta y no supera el 20%. Azitromicina, claritomicina y telitromicina no están aconsejadas para usar en hembras preñadas debido a su potencial de provocar defectos en el feto.
Con respecto a las fluoroquinolonas, no están indicadas en ningún momento de la preñez por su capacidad de reducir la osificación y provocar lesiones en el cartílago articular del feto. Se ha reportado que altas dosis producen a aborto.
Cloranfenicol está contraindicado en todas las especies por su capacidad de provocar parto prematuro acompañado del llamado síndrome del bebe gris, que refleja un colapso cardiovascular por acumulación del fármaco en los compartimientos fetales.
Las tetraciclinas afectan la osificación y el desarrollo fetal y se las asocia con hepatopatías en el feto. Solo deben utilizarse en casos donde no exista otra posibilidad de tratamiento. Si se decide la administración se aconseja no modificar la dosis.
Consideraciones finales
La terapéutica de las hembras preñadas es un desafío terapéutico debido a las modificaciones fisiológicas y bioquímicas de la preñez que conducen a cambios en la farmacocinética de los fármacos administrados. Ante la llegada de una hembra preñada al consultorio, no se deben olvidar las preguntas clave: ¿Es imprescindible tratarla farmacológicamente? ¿La patología permite (aplicando mediadas de contención) retrasar el inicio del tratamiento hasta el parto?.
De ser imprescindible el tratamiento, se deben buscar las opciones más seguras sin olvidar que todas ellas esconden un riesgo. Sin embargo, el conocimiento de las consecuencias que los cambios fisiológicos de nuestra paciente tendrán en el comportamiento farmacocinético del fármaco seleccionado reducirá de manera significativa los riesgos e incrementará el éxito terapéutico.

