










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales (Asociación Física Argentina)
versión impresa ISSN 0327-358Xversión On-line ISSN 1850-1168
An. AFA vol.28 no.3 Tandil set. 2017
BIOFÍSICA Y FISICA MÉDICA
Estudio por dinámica molecular de la hidratación de la proflavina en solución
Study for molecular dynamics of proflavin hydration in solution
M. G. Campoa, G.M. Corrala*
a Facultad de Ciencias Exactas y Naturales – Universidad Nacional de La Pampa – Uruguay 151 – (6300) La Pampa – Argentina
email: mario@exactas.unlpam.edu.ar
Resumen
Se estudia la hidratación de la proflavina doblemente protonada (C13N3H13) en solución mediante dinámica molecular clásica. Se utiliza el paquete de simulación GROMACS. La topología de las moléculas se basa en el campo de fuerzas Amber99, y como solvente se usa el agua SPC/E. Se analiza el comportamiento de la proflavina mediante el estudio de las funciones de distribución radial, establecimiento de puentes de hidrógeno entre átomos de la proflavina y aguas que la rodean, y distribuciones de frecuencia de ocurrencia de puentes de hidrógeno en el agua. Se halla que los grupos amino estructuran el agua que los rodean formando frecuentemente puentes de hidrógeno, mientras que esta estructura se adapta a la del solvente. La estructura aproximadamente tetraédrica del agua es afectada por la presencia de la proflavina en la primer capa de hidratación (0,25 nm) y en dos regiones del solvente de 0,15 nm de espesor que rodean a la proflavina por encima y por debajo del plano de los anillos, donde la frecuencia de ocurrencia de 4 puentes de hidrógeno disminuye entre un 25 % y un 50 %.
Palabras clave: Proflavina; Dinámica molecular; Puentes de hidrógeno; Hidratación.
Abstract
The hydration of a doubly protonated proflavine (C13N3H13) in solution is studied by classical molecular dynamics. The GROMACS simulation package is used. The topology of the molecules is based on the Amber99 force field, and SPC/E water model is used as solvent. The proflavine behavior is analyzed by the study of the functions of radial distribution, the establishment of hydrogen bonds between proflavine atoms and surrounding water molecules, and the distributions of frequency of occurrence of hydrogen bonds in water. It is found that the amino groups structure the water that surround them often forming hydrogen bonds, whereas this structure is adapted to that of the solvent. The approximately tetrahedral structure of the water is affected by the presence of proflavine in the first hydration layer (0.25nm) and in two region of the solvent with 0.15 nm of thickness surrounding the proflavine above and below the plane of the rings, where the frequency of occurrence of 4 hydrogen bonds decreases between 25% and 50 %.
Keywords: Proflavine; Molecular dynamics; Hydrogen bonds; Hydration.
Recibido: 26/02/17;
aceptado:15/06/17
I. INTRODUCCIÓN
La molécula de proflavina, al igual que el conjunto de los derivados de la acridina, se intercala y fija externamente en el ADN, afectando así muchos procesos biológicos como la replicación y la transcripción. Así, resulta relevante su estudio para el diseño de fármacos con el fin de desarrollar agentes quimioterapéuticos contra diferentes tipos de cáncer, malaria, bacterias y virus (entre otros)1-6. Su estructura plana de tres anillos es de importancia crítica en tales procesos de intercalación, mientras que sus dos grupos aminos contribuyen a la interacción electrostática con los grupos fosfato del ADN7.
Una de las características observables de la proflavina es su agregación en solución, formando dímeros a concentraciones mayores a 10-5 M. Además, se encuentra que dichas moléculas se apilan manteniendo entre ellas una distancia de 3,4 Å, y forman grandes agregados a concentraciones mayores a 10-2 M 8-10.
Estudios previos de difracción de rayos X y Density Functional Theory (DFT) muestran que los grupos amino de la proflavina son hidrofílicos10,11. Además, se manifiesta un comportamiento hidrofóbico de los grupos CH de la molécula cuando está intercalada o fijada al ADN12.
La interacción hidrofóbica resulta de mayor importancia que la electrostática en el proceso de fijación e intercalación de la proflavina en el ADN13. Durante la intercalación se produce el rompimiento de puentes de agua internos entre átomos de ADN, lo que disminuye su estabilidad térmica14.
La estructura y dinámica del agua, y en particular de los puentes de hidrógeno, se ha investigado utilizando tanto técnicas experimentales como teóricas lográndose una extensa caracterización15-17. El modelo de agua SPC/E ha demostrado fiabilidad en la representación del agua cuando se lo utiliza en dinámica molecular18,19.A temperatura ambiente cada molécula de agua SPC/E establece aproximadamente 4 puentes de hidrógeno con las aguas que la rodean cuyo tiempo de vida medio es del orden del ps, estableciendo de esa manera una estructura aproximadamente tetraédrica18-22. Por otro lado, diferentes estructuras del agua cumplen roles relevantes en procesos de interacción de moléculas en solución. Por ejemplo en la superficie del ADN, el agua conforma cadenas o “espinazos” de hidratación en sus gargantas menor y mayor23-26.
La intercalación y fijación de la proflavina en el ADN se ha estudiado extensamente por métodos experimentales y teóricos2,8,27-34. Sin embargo, en aquellos en que se utilizaron métodos de dinámica molecular clásica, no se han estudiado los cambios estructurales del solvente en el entorno de la molécula.
En este trabajo se desarrolla un modelo de proflavina para analizar su hidratación desde el punto de vista estructural del solvente, discutiendo su posible influencia en los fenómenos de intercalación y fijación en el ADN.
II. MÉTODO
Se utilizó dinámica molecular clásica (MD) mediante el paquete de simulación GROMACS35. Se desarrolló un modelo de proflavina (Figura 1) doblemente protonada C13H13N3 (PF) a partir del campo de fuerzas AMBER9936,37, completando los parámetros de potenciales de interacción no definidos de acuerdo al protocolo previsto en la bibliografía35. Las cargas se calcularon mediante el análisis de Mulliken con el módulo DFT de NWChem38, de manera que su carga total fuese +2e. El modelo de ión cloro corresponde a la base AMBER99, mientras que para el solvente se utilizó el modelo SPC/E18.

Figura 1: Representación esquemática de la C13H13N3 con los nombres asignados a cada átomo.
Se simula una molécula de proflavina en una caja rectangular con 2 iones cloro (con lo cual la carga neta del sistema es cero) y 1410 moléculas de agua, con condiciones periódicas de contorno. Los potenciales de interacción utilizados son los usuales descritos en la bibliografía35. El protocolo de simulación fue el siguiente: (a) Iniciación: Minimización de energía del sistema, y posterior asignación de velocidades al solvente acorde a una distribución de Maxwell-Boltzmann a 300 K. (b) Estabilización 1: MD sometiendo la molécula de PF a restricción posicional y el sistema a volumen constante. (c) Estabilización 2: Continuación con una MD nuevamente con la PF con restricción posicional y el sistema a presión constante utilizando un barómetro de Parrinello-Rahman. En ambos pasos (b) y (c) se utilizó como criterio de corte para dar por terminado el proceso de estabilización, el que el sistema alcance una energía potencial constante, caso en el cual se consideró que el sistema había alcanzado el equilibrio. (d) MD de estabilización: MD sin restricciones posicionales usando un termostato de Berendsen modificado con V-rescale, y un barómetro de Parrinello-Rahman a 1 atm y 300 K. (e) MD para el análisis de datos de 40 ns con el mismo baño hidrostático.
Se analiza el comportamiento de la función de distribución radial (FDR) de átomos entorno de otrosátomos, la cual es una herramienta usual en experimentos, teoría y simulación, para caracterizar el estado de la materia condensada39. Una FDR da la probabilidad de hallar un determinado átomo a una distancia r de otro, relativa a la probabilidad esperada para una distribución completamente aleatoria con la misma densidad numérica.
Usando las FDR se obtiene el número de hidratación N, que resulta el número de moléculas de agua en la primer capa de hidratación:
Siendo □ la densidad numérica de oxígenos de agua, g(r) la FDR, y rmin la posición del primer mínimo de la función g(r) .
Se utiliza una definición geométrica de puentes de hidrógeno, considerando 3,5 Å como distancia máxima entre donor y aceptor, y un ángulo mínimo de 145° para el enlace Donor – H – Aceptor40,41. El concepto de puente de hidrógeno se usa para estudiar dos conjuntos de casos diferentes. Por un lado se considera (a) El porcentaje de puentes de hidrógeno (%HB) en átomos específicos de la PF, que indica con qué frecuencia se encuentra en la simulación el establecimiento de un puente de hidrógeno que tiene a un átomo de la molécula como donor (carbono o nitrógeno) y un oxígeno de agua como aceptor. Por el otro, se analiza en zonas específicas de interés del solvente la distribución de frecuencias de ocurrencia de puentes de hidrógeno HB(n) (n=1,..,7) entre moléculas de agua entre sí. Los valores de las HB(n) (que oscilan entre 0 y 1), representan la frecuencia con que se observa (en promedio) en la simulación, que una molécula de agua establece n puentes de hidrógeno con aquellas que la rodean.
Se distinguen 3 capas de solvente alrededor de la PF para el análisis de las HB(n) y, a su vez, 2 zonas en cada una de las capas. En la primer capa de solvente se distingue, por un lado, una zona que “rodea” los anillos de la PF que denominamos L1, constituida por las moléculas de agua donde el ángulo entre el plano del anillo más cercano y la posición del oxígeno del agua relativa al carbono o nitrógeno más cercano de la PF se encuentre en el intervalo > >− (ver figura 2). Por otro lado, queda determinada una segunda zona T1 constituida por moléculas de agua cuyos oxígenos no cumplen la condición anterior. Esto permite analizar si la presencia de la proflavina afecta de manera diferenciada la estructura del agua que se encuentra en la zona lateral de la primer capa (L1), de aquellas que se encuentran en las zonas aproximadamente paralelas a los anillos, por encima y por debajo del plano que forman los mismos (T1). De manera análoga se definen las zonas L2 , T2 y L3, T3, para dos capas de solvente adicionales que envuelven a la proflavina.
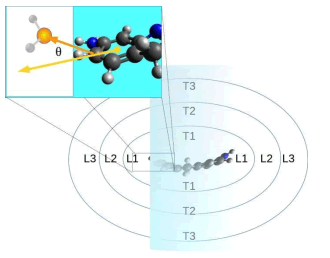
Figura 2: Esquema representativo de la división en zonas ocupadas por moléculas de agua que rodean a la proflavina para el análisis de las distribuciones de frecuencia de ocurrencia de puentes de hidrógeno entre aguas. Ver el texto para los detalles.
III. RESULTADOS
La figura 3 muestra FDR de oxígenos de agua alrededor de algunos hidrógenos de la proflavina (gHOw(r)). A su vez, en la Tabla 1 se detallan algunas características de FDRs relevantes para el análisis. Dada la simetría de las molécula de proflavina, se promedian los resultados de aquellas funciones que se consideran (y se verifica que resultan) equivalentes, por ejemplo la gHaminosOw(r) correspondiente a H13, H14; H25 y H24.
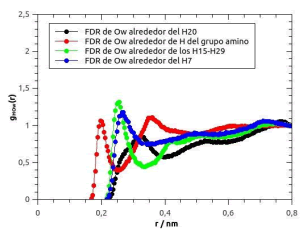
Figura 3: gHOw(r) de oxígenos de agua alrededor de hidrógenos de la proflavina. Ver el texto para más detalles.
TABLA 1: CARACTERÍSTICAS DE LAS FUNCIONES DE DISTRIBUCIÓN RADIAL


Considerando la posición del primer pico en la correspondiente gHOw(r), el mayor acercamiento de oxígenos de agua (Ow) a la proflavina se produce en hidrógenos de los grupos aminos (H13,14, H24,25). Se observa una estructura de hidratación bien definida y más compacta que el resto de la molécula, teniendo en cuenta la posición del primer mínimo de la misma función. La existencia de un segundo pico bien definido indica la posible formación de una segunda capa de hidratación, aunque podría deberse a la presencia de Ow rodeando al otro hidrógeno del grupo. El %HB entre el nitrógeno del grupo y los Ow que lo rodean resulta de un 63 %, lo que indica que el grupo amino logra que las moléculas de agua se estructuren fuertemente con él.
El número de hidratación mayor, y la altura de primer pico mayor ocurre en los hidrógenos H15-29 del carbono central C9 acorde con una primer capa de hidratación poblada. Sin embargo, puede observarse que esto se debe a que la posición del primer pico (0,24) y del primer mínimo (0,33) es mayor que en el caso de la gHaminosOw(r) de los hidrógenos del grupo amino, con lo cual la hidratación de esta zona resulta menos compacta, siendo el volumen abarcado (y por lo tanto la cantidad de moléculas de agua) también mayor. Sin embargo, tanto el bajo %HB con moléculas de agua de la primer capa (0,15 %), y las no existencia de un segundo pico bien definido en la gH15-29Ow(r), indica que el solvente prácticamente no se estructura con esta región de la molécula.
Para el resto de los hidrógenos ligados a carbonos de la proflavina se observa en general un comportamiento de la gHOw(r) similar al que ocurre en los hidrógenos del C9, con algunos matices. En general se observa una poca estructuración del solvente alrededor de los mismos dados los %HB bajos, y una capa de hidratación que los rodea muy difusa, es decir, primeros picos y primeros mínimos de la gHOw(r) menos definidos y en posiciones alejadas comparadas con aquellas de los hidrógenos de los aminos.
En el hidrógeno ligado al nitrógeno central se observa un pico de la FDR muy alejado (0,33 nm) y valores de la función menores a 1 para r< 0,7 nm, asociable a una pobre hidratación de la molécula en dicha zona.
Para el análisis de la la frecuencia de ocurrencia de puentes de hidrógeno entre aguas HB(n) se consideran espesores de 0,25 nm, 0,15 nm, y 0,2 nm para la primera, segunda y tercer capa de solvente alrededor de la PF, respectivamente.
La distribución de frecuencias de ocurrencia entre aguas en la primer capa de solvente que rodea a la PF muestra un máximo en HB(3) (tanto en la zona L1 como T1), mostrando que la estructura tetraédrica que se observa en el bulk del solvente, que está asociada a una frecuencia de ocurrencia mayor de 4 puentes de hidrógeno, se ve fuertemente disminuida (~25%) por la presencia de la molécula de proflavina (ver Figura 4). Significativamente, este hecho se repite sólo en la zona T2 la segunda capa de solvente. La distribución HB(n) en la tercer capa de hidratación de la PF es similar al del bulk, indicando que estructura del agua en esta región no es afectada por la presencia de la molécula. Dado que las zonas T1 y T2 son paralelas a los anillos de la proflavina, se deduce que la presencia de esta molécula afecta más a la estructura del solvente en esta dirección, prevaleciendo una estructura de 3 puentes de hidrógeno (HB(3)~0,45) entre moléculas de agua que se encuentran rodeando a la PF por encima y por debajo de ella.
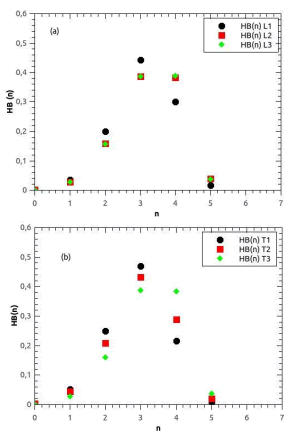
Figura 4: Distribuciones de frecuencia de ocurrencia de puentes de hidrógeno entre moléculas de agua. (a) zonas de hidratación laterales sucesivas L1, L2 y L3. (b) zonas
IV. DISCUSIÓN Y CONCLUSIONES
El modelo de molécula de proflavina que se ha desarrollado y utilizado para la simulación por el método de dinámica molecular clásica, verifica un conjunto de comportamientos de esta molécula en solución ya observados en investigaciones previas10-14. A su vez, se obtiene una caracterización fehaciente de su hidratación, que podrían permitir explicar algunos comportamientos de la proflavina y su interacción con el ADN.
En primer lugar, si bien en la bibliografía ya se indica que los grupos amino son proclives a establecer enlaces con el agua que los rodean10,11 en este trabajo se observa además que estos enlaces se constituyen frecuentemente en puentes de hidrógeno. La estructuración mediante la formación de puentes de hidrógeno del solvente alrededor de los grupos mencionados afecta la estructura del agua sólo en la primer capa de hidratación.
En segundo lugar, mediante el estudio realizado se obtienen detalles significativos del comportamiento hidrofóbico de la proflavina ya señalado en la bibliografía12. Por un lado, hay una escasa presencia de moléculas de agua en el entorno cercano al nitrógeno central y a los carbonos con un único hidrógeno ligado, y en general se observa que la frecuencia con que estosátomos establecen puentes de hidrógeno con las moléculas de agua de su entorno es pequeña, aspectos usualmente asociados a un comportamiento hidrofóbico. Además, la estructura aproximadamente tetraédrica del agua es afectada por la presencia de la proflavina en la primer capa de hidratación (∼0,25 nm dos regiones del solvente de la proflavina (∼0,15 nm de espesor), que la envuelven por encima y por debajo paralelas a los planos de sus anillos (ver Figura 5). En estas regiones la frecuencia de ocurrencia de 4 puentes de hidrógeno di un 25 % y un 50 %, prevaleciendo una estructura con 3 puentes de hidŕogeno. Llamativamente el espesor de esta zona del solvente afectada por encima y por debajo de la molécula coincide aproximadamente con el espacio de separación entre moléculas de proflavina durante la formación de apilamientos en la agregación8-10, lo que podría indicar que este fenómeno estaría dominado por una interacción hidrofóbica.

Figura 5: Esquema donde se indican en celeste las regiones del solvente paralelas a los anillos de la proflavina (∼0,4 nm de espesor) donde la presencia de la molécula} “rompe” la estructura aproximadamente tetraédrica del agua. Ver texto para más detalles.
Posiblemente, el “rompimiento” de la estructura del agua por encima y por debajo del plano de los anillos de la proflavina facilite la intercalación entre las bases del ADN, considerando que se trata de un proceso dominado por una interacción hidrofóbica. La desestructuración del solvente se condice con lo señalado por Kostjukov el al.14, quien asocia la disminución de la estabilidad térmica del ADN en presencia de proflavina, indicando que la intercalación de esta última molécula rompe puentes de agua preexistentes entre las bases.
Agradecimientos
M. G. Campo y G. M. Corral agradecen e financiamiento provisto por PICTO UNLPAM 2005 30807 y la Facultad de Ciencias Exactas de la UNLPam
1. Lerman, L. S. Structural considerations in the interaction of DNA and acridines. J. Mol. Biol., 3, 18-30 (1961). [ Links ]
2. Li, H. J. and Crothers, D. M., Relaxation studies of the proflavine-DNA complex: the kinetics of an intercalation reaction. J. Mol.Biol., 39, 461-& (1969). [ Links ]
3. Ngadi, L., Galy, A., Galy, J., Barbe, J., Crémieux, A., Chevalier, J., Sharples, D. Some new 1-nitro acridine derivatives as antimicrobial agents. Eur. J. Med. Chem., 25, 67–70 (1990).
4. Chacón-García, R. M. a L. The search of DNA-intercalators as antitumoral drugs: what it worked and what did not work. Curr. Med. Chem., 12, 127 151 (2005). [ Links ]
5. Pal Singh, N., Kumar, R., Prasad, D.N., Sharma, S., Silakari, O. Nutrition in pregnancy: the argument for including a source of choline. Int. J. Biol. Chem. 5, 193–199 (2011).
6. Belmont, P., Bosson, J., Godet, T., Tiano, M. Acridine and acridone derivatives, anticancer properties and synthetic methods: where are we now?. Anticancer Agents Med. Chem., 7, 139–169 (2007).
7. Nakamoto, K., Tsuboi, M., Strahan, G.D. Intercalating drugs, in: Drug–DNA Interactions, John Wiley & Sons, Inc., 119–208 (2008).
8. Neidle, S. and Jones, T. A. Crystal structure of proflavine, a DNA binding agent. Nature 253, 284-285 (1975). [ Links ]
9. Haugen, G. R. and Melhuish, W. H. Association and self-quenching of proflavine in water. Transactions of the Faraday Society 60, 386-394 (1964). [ Links ]
10. Obendorf, S. K., Carrell, H. L. and. Glusker, J. P. Enhancing channelrhodopsins: an overview. Acta Crystallographica Section B, 30, 1408-1411 (1974). [ Links ]
11. Qin, Z., Li, X., Zhou, M., J. Chin. Chem. Soc. 61, 1-6 (2014). [ Links ]
12. Kim, K.S., Corongiu, G., Clementi, E. Networks of water molecules in a proflavine deoxydinucleoside phosphate complex. J. of Biomol. Struct. and Dyn., 1, 263-285 (1983). [ Links ]
13. García, B., Leal, J. M., Ruiz, R., Biver, T., Secco, F. and Venturini, M., Solvent Effects on the Thermodynamics and Kinetics of Coralyne Self-Aggregation. J. Phys. Chem. B, 114, 8555–8564 (2010).
14. Kostjukov, V. V., Lantushenko, A.O., Davies, D.B., Evstigneev,M.P. On the origin of the decrease in stability of the DNA hairpin d(GCGAAGC) on complexation with aromatic drugs. Biophys. Chem., 129, 56– 59 (2007).
15. Narten, A. H., Danford, M. D. and Levy, H. A. Copper dioxygen (bio)inorganic chemistry. Faraday Discuss., 43, 97-108 (1967). [ Links ]
16. Soper, A.K., Bruni, F. and Ricci, M. A. Aromatic reactivity. Part XIII. Effects of methyl and t-butyl groups in aromatic detritiation. J. Chem. 106, 247-254 (1997). [ Links ]
17. Modig, K., Pfrommer, B. G. and Halle, B. Temperature-dependent hydrogen-bond geometry in liquid water. Phys. Rev. Lett., 90(7), 075502 (2003). [ Links ]
18. Berendsen,H. J. C. , Grigera, J. R. and Straatsma, T. P. The missing term in effective pair potentials. J. Phys. Chem., 91, 6269-6271 (1987). [ Links ]
19. Chatterjee, S., Debenedetti, P. G., Stillinger, F. H. and Lynden-Bell, R. M. A computational investigation of thermodynamics, structure, dynamics and solvation behavior in modified water models. J. Chem. Phys., 128, 124511 (2008). [ Links ]
20. Mark, P., & Nilsson, L. J. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. Phys.Chem. A, 105(43), 9954-9960 (2001). [ Links ]
21. Campo, M. G., Ferri G. L., Roston G. B. J. Phys.,39,Braz. 439-443 (2009). [ Links ]
22. Campo, M. G., Structural and dynamic properties of SPC/E water. Papers in Phys.,2, 020001 (2010). [ Links ]
23. Young, M. A., Jayaram, B. and Beveridge, D. L. Intrusion of Counterions into the Spine of Hydration in the Minor Groove of B-DNA: Fractional Occupancy of Electronegative Pockets. J. Am. Chem. Soc., 119, 59–69 (1997).
24. Denisov,V. P., Carlstrom, G. , Venu, K. and Halle, B. Kinetics of DNA hydration. J. Mol. Biol., 268, 118–136 (1997).
25. Bonvin, A. M. J., Sunnerhagen, M., Otting, G., van Gunsteren, W. F. Water molecules in DNA recognition II: a molecular dynamics view of the structure and hydration of the trp operator. J. Mol. Biol., 282, 4, 859–873 (1998).
26. MacKerell, A. D., Banavali, N. K., All-atom empirical force field for nucleic acids: 2) Application to molecular dynamics simulations of DNA and RNA in solution. J. Comp. Chem., 21, 2, 105-120 (2000). [ Links ]
27. Ramstein, J., Ehrenberg M. and Rigler, M. Biochemistry (Mosc.) , 19, 3938-3948 (1980). [ Links ]
28. Bereznyak, E., Gladkovskaya, N., Khrebtova, A. Dukhopelnikov, E. and Zinchenko, A. Peculiarities of DNA-proflavine binding under different concentration ratios. Biophysics, 54, 574-580 (2009). [ Links ]
29. Tang, P., Juang, C. L. and Harbison, G. S. Intercalation complex of proflavine with DNA: structure and dynamics by solid-state NMR. Science, 249, 70-72 (1990). [ Links ]
30. Pack, G. R., Hashimoto, G. M. and Loew, G. H. Ann. N. Y. Quantum chemical calculations on the two-step mechanism of proflavin binding to DNA. Acad. Sci. 367, 240-249 (1981). [ Links ]
31. Dearing, A., Weiner, P. and Kollman, P. A. Nucl. Acids Res.,9 , 1483-1498 (1981 ). [ Links ]
32. Sasikala, W. D. and Mukherjee, A. Single water entropy: hydrophobic crossover and application to drug binding. J. Phys. Chem. B, 116, 12208-12212 (2012). [ Links ]
33. Sasikala, W. D. and Mukherjee, A. Intercalation and de-intercalation pathway of proflavine through the minor and major grooves of DNA: roles of water and entropy. Phys. Chem. Chem. Phys., 15, 6446-6455 (2013). [ Links ]
34. Sasikala, W. D. and Mukherjee, A. Structure and dynamics of proflavine association around DNA. Phys. Chem. Chem. Phys., 18(15), 10383-10391 (2016). [ Links ]
35. Van Der Spoel, D., Lindahl, E., Hess, B., Van Buuren, A.R., Apol, E. et al. Gromacs user manual , version 5.4, disp. en http://www.gromacs.org/Documentation/Manual (accedido el 20/02/2017). [ Links ]
36. Sorin, E. J. and Pande, V. S. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophysical Journal, 88, 2472-2493 (2005). [ Links ]
37. DePaul A. J., Thompson E. J., Patel S. S., Haldeman K. & Sorin E. Equilibrium conformational dynamics in an RNA tetraloop from massively parallel molecular dynamics. J.Nucleic Acids Research, 38, 4856-4867 (2010). [ Links ]
38. Valiev, M., Bylaska, E. J., Govind, N., Kowalski, K., Straatsma, T. P., Van Dam, H. J., ... & De Jong, W. A. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Computer Physics Communications, 181(9), 1477-1489 (2010). [ Links ]
39. Allen, M. P., Tildesney, D. J. In “Computer Simulation of Liquids”, Clarendon, Ed., Oxford, (1987).
40. Luzar A. and Chandler D. Hydrogen-bond kinetics in liquid water. Nature, 379, 55-57, (1996a). [ Links ]
41. Luzar A. and Chandler D. Effect of environment on hydrogen bond dynamics in liquid water. Phys. Rev. Lett., 76, 928-931, (1996b). [ Links ]

