











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkI. INTRODUCCIÓN
Las celdas de combustible hidrógeno-oxígeno brindan una solución energética indiscutible debido a su alta eficiencia, emisiones nulas, proceso silencioso y fuente renovable prácticamente ilimitada de reactivos1-4, pero todavía hay muchos aspectos a mejorar. Entre los procesos que ocurren en la superficie de los electrodos de las celdas a hidrógeno en general y las de electrolito de membrana polimérica PEMFC, en particular, hay dos que son los más importantes: la oxidación de hidrógeno (ROH) en el ánodo y la reducción de oxígeno (RRO) en el cátodo. Mientras que la ROH tiene un sobrepotencial de oxidación bajo y una cinética rápida, la RRO presenta una reacción electroquímica muy lenta, característica de varias rutas de reacción diferentes, dirigida principalmente por la acción catalítica del soporte metálico y por el pH de la solución5-8. Esta reacción afecta en mayor medida el desempeño de la celda y conduce a un sobrepotencial más alto en la condición de operación de una PEMFC. Por ello las investigaciones están orientadas a disminuir el sobrepotencial para la RRO y mejorar su cinética.
El oxígeno, como especie preadsorbida, puede reducirse directamente a agua, en medios ácidos o en iones OH, en medios alcalinos, consumiendo cuatro electrones por una molécula de O2, o indirectamente, formando un producto intermedio, ya sea peróxido de hidrógeno, en ácidos, o iones  en medios alcalinos, cuando solo se consumen dos electrones por una molécula de O2. Obviamente la reducción directa es más eficiente. Por ello es necesario lograr nuevos electrocatalizadores que eviten la formación de intermediarios9.
en medios alcalinos, cuando solo se consumen dos electrones por una molécula de O2. Obviamente la reducción directa es más eficiente. Por ello es necesario lograr nuevos electrocatalizadores que eviten la formación de intermediarios9.
La tendencia actual es lograr la disminución de la cantidad de metales nobles en los electrodos catalíticos, siendo esto un reto importante en el desarrollo de celdas de combustible cada vez más económicas10-12. Tal reducción en la cantidad de catalizador, en la mayoría de los casos, se puede conseguir aumentando el área activa del metal que se utilice sobre la superficie del electrodo10,11. Una forma de lograr esto es el desarrollo de nuevos soportes y recubrimientos que aumenten la durabilidad de la capa catalítica, lo que generalmente permite una mayor utilización del catalizador metálico12.
La cinética y el mecanismo de la RRO fueron ampliamente investigados principalmente en platino, conocido como el mejor electrocatalizador ya que admite la reducción de O2 en cuatro electrones a sobrepotenciales relativamente bajos5-7,13-17. Pero como este metal es muy escaso y oneroso, con el fin de bajar los costos, se invirtió mucho esfuerzo para mejorar su eficacia catalítica, ya sea dispersándolo sobre un soporte de carbono de alta área superficial18,19, o aleándolo con metales menos nobles19. Además, los catalizadores de platino utilizados actualmente en celdas de combustible de baja temperatura sufren una pérdida continua de actividad, causada por una fuerte adsorción de CO presente a nivel de impureza en el gas combustible.
El oro, por el contrario, es un electrocatalizador mucho menos activo para la RRO en medios ácidos, y en ese caso se produce una reducción de oxígeno de dos electrones en los electrodos de Au, originando el peróxido de hidrógeno, que se reduce solamente a altos sobrepotenciales20-24. Sin embargo, Haruta et al.25 publicaron que los nanoclusters de Au dispersados en un soporte de alúmina muestran una actividad inesperadamente alta hacia la oxidación de CO26 y la reducción de los óxidos de nitrógeno27. El descubrimiento de que el oro nanodispersado permite la oxidación a baja temperatura del CO, muy interesante por el problema de las celdas de combustible de óxido sólido, apuntado anteriormente, hace presuponer que puede ser importante para mejorar otros procesos en electrocatálisis.
La mejora mutua de la actividad catalítica del catalizador metálico y el material de soporte se denominó interacción fuerte de soporte metálico (IFSM o SMSI, por sus siglas en inglés)25-27. Este efecto se observó mucho antes en el sistema de titanio/platino28 y, por lo tanto, desde el punto de vista de la IFSM, el titanio fue considerado hace mucho tiempo como un material de soporte muy prometedor para la catálisis y la electrocatálisis. Eso estimuló a muchos autores a investigar el comportamiento electroquímico de las películas compuestas de metal/titanio sintetizadas de varias maneras28-40. Para ello, películas compuestas de diferentes óxidos metálicos se aplicaron sobre la superficie de titanio principalmente por descomposición térmica de compuestos inestables de titanio y metales nobles o de transición con el objeto de estudiar su actividad catalítica25-33.
La película de óxido formada espontáneamente sobre el titanio lo pasiva, haciéndolo resistente a la corrosión. La aplicación de corriente anódica incrementa el espesor del óxido, el cual está determinado por el potencial final. Al titanio se lo denomina metal válvula, debido a que no permiten el pasaje de corriente en ambas direcciones. Este efecto de rectificación se interpreta de dos maneras: 1. La corriente catódica es casi cero, y una corriente anódica es posible cuando el potencial excede aproximadamente el 50% del potencial de formación. Esto es cierto para capas gruesas de óxido y, 2. La corriente anódica es baja para potenciales inferiores al potencial de formación, pero es posible una fuerte evolución de hidrógeno (catódica)41. Este óxido es amorfo a temperatura ambiente y tiene un espesor de 1 a 6 nm42. La película de óxido pasiva, compuesta de dióxido de titanio (TiO2), puede ser formada por oxidación térmica o por anodización, y presenta características de un semiconductor. Durante la oxidación anódica diferentes tipos de óxidos de Ti (TiO, TiO2, Ti2O3 y Ti3O5) se forman sobre la superficie del Ti, donde el TiO2 es la película de óxido más estable y la que se encuentra con mayor frecuencia. Las propiedades del óxido dependen del método de preparación y de la técnica que se utiliza para formar las películas de óxido en metales válvula43.
Baez y Pletcher44 compararon el comportamiento de las capas compuestas de Au/TiO2 producidas por varias formas diferentes previamente descritas en la literatura. Dado el potencial muy negativo del titanio puro, la corrosión rápida comienza inmediatamente después de su inmersión en cualquier solución acuosa. En soluciones acuosas moderadamente ácidas, neutras y moderadamente alcalinas, según los diagramas de Pourbaix45, el TiO2 es el óxido de titanio más estable en soluciones acuosas, y la corrosión puede describirse mediante la ecuación:
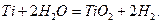 (1)
(1)
Este comportamiento del titanio permite también la deposición de más metales nobles a partir de las soluciones de sus sales, por reacción de sustitución34. La resistividad de la capa de TiO2 es bastante alta, alrededor de 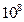
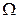 cm 46.
cm 46.
Metales como el Au y Ag, en determinadas condiciones, pueden mostrar una efectividad en la reducción de oxígeno cercana a la del platino47-49. Se encontró que la RRO se desarrolla sobre varias formas de dióxido de titanio, aunque a una sobretensión mucho mayor en comparación con la del platino. Clechet et al. evidenciaron que esta reacción tiene lugar en una capa de TiO2 formada galvanostáticamente50. Más recientemente, Clark et al. publicaron que en el TiO2, formado anódicamente, la sobretensión para la RRO puede reducirse significativamente después de una activación extensa de la capa de óxido por polarización cíclica dentro de un rango de potencial limitado51.
La voltamperometría cíclica proporciona en forma rápida información cualitativa sobre los catalizadores y las reacciones electroquímicas. El potencial de inicio ( ) de la RRO y la corriente máxima demuestran la actividad catalítica de un catalizador para esta reacción52.
) de la RRO y la corriente máxima demuestran la actividad catalítica de un catalizador para esta reacción52.
Los datos en literatura muestran que los estudios de electrodos de óxido de titanio en solución ácida y alcalina presentan actividad catalítica hacia la RRO y son dependientes del pretratamiento de la superficie sobre la cual se crece el óxido anódico31,50,53-56.
El objetivo de este trabajo es el estudio de la RRO sobre los electrodos con deposición potenciostática de Au, antes y después del crecimiento del óxido electroquímico (OEQ) a diferentes potenciales finales, a fin de determinar las mejores condiciones de preparación de los sustratos para una mejor electrocatálisis.
II. PARTE EXPERIMENTAL
Se utilizaron 6 (seis) electrodos de trabajo idénticos, cada uno de ellos formado por una lámina de titanio grado 1 (99.9%) de 10 mm 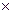 10 mm de área (con una solapa de contacto de 0.7 mm) y 0.3 mm de espesor.
10 mm de área (con una solapa de contacto de 0.7 mm) y 0.3 mm de espesor.
Para asegurar una superficie limpia, los electrodos de trabajo (ET) se lavaron antes de su uso con abundante agua ultrapura Milli-Q (agua desionizada, resistividad de 18.2 Mcm); alcohol etílico y acetona-etanol a temperatura ambiente, y sometidos a ultrasonido durante 7 minutos (tanto en agua como en acetona), para después generar el óxido térmico en un horno a 450ºC durante una hora y con enfriamiento gradual. Luego se realizó el pulido de una de las caras de cada electrodo, lo cual eliminó completamente el óxido térmico. Este pulido consistió en el lijado unidireccional con lijas de diferentes tamaños de grano, desde 320 hasta 2500, y finalmente con alúmina 1 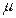 m hasta lograr una superficie espejada. Posteriormente se volvió a realizar la rutina de limpieza con agua Milli-Q, alcohol y acetona en ultrasonido. En todos los ET el contacto fue recubierto con un soporte de teflón dejando expuesto solo el área de trabajo. El contra electrodo (CE) utilizado fue una hélice de Pt de 3 mm de diámetro y 2 mm de longitud. Para conseguir superficies limpias y reproducibles de este último, se realizó un tratamiento a la llama, utilizando un mini soplete de laboratorio a gas butano.
m hasta lograr una superficie espejada. Posteriormente se volvió a realizar la rutina de limpieza con agua Milli-Q, alcohol y acetona en ultrasonido. En todos los ET el contacto fue recubierto con un soporte de teflón dejando expuesto solo el área de trabajo. El contra electrodo (CE) utilizado fue una hélice de Pt de 3 mm de diámetro y 2 mm de longitud. Para conseguir superficies limpias y reproducibles de este último, se realizó un tratamiento a la llama, utilizando un mini soplete de laboratorio a gas butano.
El electrodo de referencia (ER) utilizado en las mediciones electroquímicas fue el de calomel saturado (ECS, E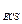 = +0.244 V vs ENH).
= +0.244 V vs ENH).
Las soluciones de trabajo fueron preparadas con reactivos para análisis (Merck), usando también agua ultra-pura Milli-Q, que fue obtenida de un sistema Elix de la compañía Milli-Q.
Para la electrodeposición de Au se utilizó una solución de HAuCl4H2O 0.01 M disuelto en HCl 0.1 M y para los estudios electroquímicos (EEQ) en una solución 0.01 M HClO4.
En todos los casos, las soluciones se desoxigenaron mediante burbujeo continuo de nitrógeno de alta pureza (99.999%, AGA) y, para el EEQ del oxígeno, las soluciones se saturaron con oxígeno (99,98%, AGA).
Se utilizó una celda de vidrio de borosilicato del tipo convencional, con tres entradas para los correspondientes ET, CE y ER, y una entrada adicional para el paso del gas a través de la solución. El ER se mantuvo en un compartimento separado de la mayor parte del electrolito por un capilar de Luggin, relleno con el mismo electrolito.
Previo a cada serie de experimentos se realizaron procedimientos rigurosos de limpieza. La celda electroquímica fue introducida en solución sulfonítrica durante 24 horas y enjuagada repetidas veces con agua ultra-pura. En todos los casos, la celda electroquímica fue colocada en agua a punto de ebullición y lavada y enjuagada cuidadosamente con agua ultra pura previamente a cada medición.
Las técnicas empleadas en este trabajo son: la cronoamperometría para la deposición de Au sobre Ti y la voltamperometría cíclica (VC), para el crecimiento de los óxidos y la determinación de los parámetros electroquímicos.
En todos los casos, los experimentos se realizaron con un Potenciostato-Galvanostato Metrohm-Autolab, modelo PGSTAT302/302N controlado por computadora y equipado con el módulo Staircase y SCAN-GEN que utiliza el software Nova 2.1.4 Build 6899 Copyriht 2018 de la misma compañía. Todos los experimentos se ejecutaron a temperatura ambiente y las curvas de los VC fueron registradas a diferentes velocidades de barrido (50 mV/s y 10 mV/s) y en dirección negativa.
La preparación de los sistemas siguió dos procedimientos:
Procedimiento 1: El crecimiento de óxido y su estabilización se realizaron previamente al depósito de Au en 3 electrodos diferentes (sistema Ti/TiO2/Au).
Procedimiento 2: Se realizó primero el depósito de Au previo al crecimiento de óxido y la estabilización, en otros 3 electrodos (sistema Ti/Au/TiO2).
Los VC para el crecimiento de óxido se realizaron entre un potencial inferior de -0.3 V y un potencial superior de 1 V; 1.25 V y 1.5 V vs ECS, sobre los electrodos (cada uno a su respectivo potencial final), y la estabilización se efectuó durante 15 minutos empleando una velocidad de barrido de potencial de 50 mV/s. Para los depósitos de Au sobre Ti se utilizó la técnica de cronoamperometría a potencial de -0.6 V vs ECS, durante 10 s, previo desaireado de la solución burbujeando N2 durante 20 minutos.
Los EEQ se realizaron mediante VC a una tasa de polarización de 50 mV/s y 10 mV/s entre un potencial inicial fijo de -0.3 V, y un potencial anódico variable, de acuerdo al crecimiento de óxido que tenía cada ET, es decir de 1 V; 1.25 V y 1.5 V vs ECS.
III. RESULTADOS Y DISCUSIÓN
Comportamiento de los sistemas en presencia de N
La Fig. 1 muestra, a modo ilustrativo, un par de VC de los electrodos de Ti con Au y Ti/TiO2 (blanco), saturados en N2, preparados, respectivamente, mediante los procedimientos 1 y 2; a modo de referencia se incluye el VC del Au policristalino en la misma solución y con los mismos potenciales. Los demás electrodos presentan características similares a las mostradas en la Fig. 1.
Se puede observar que la VC del Ti/TiO2 estabilizado no presenta ningún pico en la zona anódica, mientras que la zona catódica presenta la curva característica de reducción del Ti. En la zona anódica, los VC de los sistemas Ti/TiO2/Au y Ti/Au/TiO2 presentan un comportamiento similar al VC del Au policristalino. Se pueden apreciar las cuplas redox correspondientes a la oxidación de una superficie de oro a +1.59 V y a +1.67 V. La reducción se observa alrededor de +1.16 V, en ambos casos contra ENH, lo cual es debido a la presencia de las partículas de Au.

FIG. 1: Voltamperometrías cíclicas de los sistemas obtenidos mediante el procedimiento 1 (a) y el procedimiento 2 (b).
Se puede apreciar además una mayor actividad catalítica, puesta de manifiesto por los valores de densidades de corriente, para el sistema Ti/Au/TiO2 respecto del otro sistema. La forma de los VC y los picos de potenciales, así como el comienzo de la oxidación, están en concordancia con aquellos reportados en la literatura57,58.
Electrodeposición de Au sobre los electrodos
La electrodeposición de partículas de Au se realizó en todos los ET por el método de cronoamperometría, esto es aplicando un salto de potencial constante (-0.6 V vs ECS), a tiempo de deposición () 10 s. La Fig. 2 muestra la respuesta, con uno de los ET, de la densidad de corriente (J) con el tiempo (t), luego de aplicar el salto de potencial. Los cronoamperogramas obtenidos para el resto de los ET muestran un comportamiento similar al mostrado en la Fig. 2.
Las Tablas 1 y 2 contienen la densidad de carga del Au (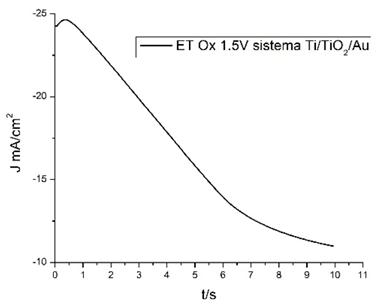 ) por unidad de área geométrica consumida durante el proceso de electrodeposición para los sistemas Ti/TiO2/Au y Ti/Au, respectivamente. La cantidad de electricidad
) por unidad de área geométrica consumida durante el proceso de electrodeposición para los sistemas Ti/TiO2/Au y Ti/Au, respectivamente. La cantidad de electricidad  (mC cm
(mC cm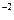 ) utilizada durante el proceso de electrodeposición a -0.6 V (vs ECS) se obtiene mediante la integral de la respuesta de densidad de corriente J vs tiempo t del cronoamperograma. Esto es, suponiendo una eficiencia de la corriente de 100% y descartando la reducción del H
) utilizada durante el proceso de electrodeposición a -0.6 V (vs ECS) se obtiene mediante la integral de la respuesta de densidad de corriente J vs tiempo t del cronoamperograma. Esto es, suponiendo una eficiencia de la corriente de 100% y descartando la reducción del H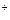 y el cargado de la doble capa, el valor de
y el cargado de la doble capa, el valor de 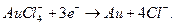 proviene solo de la siguiente reacción faradaica (si bien hay autores que afirman que la formación de Au se puede realizar en una o dos etapas, generando productos intermediarios que pueden modificar la velocidad de reacción), la cual implica 3 electrones59,60:
proviene solo de la siguiente reacción faradaica (si bien hay autores que afirman que la formación de Au se puede realizar en una o dos etapas, generando productos intermediarios que pueden modificar la velocidad de reacción), la cual implica 3 electrones59,60:
(2)
Con esto, la cantidad de Au depositado (W) en g cm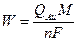 se obtiene a partir de la ley de Faraday, mediante la relación61:
se obtiene a partir de la ley de Faraday, mediante la relación61:
(3)
donde M es la masa molar del oro (196.97 gmol ), n es el número de electrones intercambiados durante el proceso de reducción (3, en este caso) y F es la constante de Faraday (96485 C mol
), n es el número de electrones intercambiados durante el proceso de reducción (3, en este caso) y F es la constante de Faraday (96485 C mol ).
).

TABLA 1: Parámetros característicos de los depósitos en los ET, con OEQ antes de la deposición de Au. ( =10 s). Sistema Ti/TiO
=10 s). Sistema Ti/TiO /Au.
/Au.
TABLA 2: Parámetros característicos de los depósitos en los ET, sin OEQ antes de la deposición de Au. (=10 s). Sistema Ti/Au.
Se puede observar que, bajo las condiciones de nuestro tratamiento experimental, los electrodos del sistema Ti/TiO2/Au (Tabla 1) presentan un mayor valor de carga consumida durante el proceso de deposición y, correspondientemente, una mayor masa de oro depositado por unidad de superficie W, respecto del sistema Ti/Au (Tabla 2). Asimismo, se observa una tendencia creciente de los valores a medida que tienen mayor crecimiento de óxido. Esto obedece bien a la ley de crecimiento de campos altos62,63.
Por otra parte los ET del sistema Ti/Au tienen valores muy similares entre sí, tanto de W como de Q, con una desviación estándar muy pequeña, lo que da cuenta de la reproducibilidad de los resultados, consecuencia de que los tres ET se prepararon de manera idéntica.
Baez y Pletcher44 encontraron que se pueden preparar recubrimientos a base de oro estables y adhesivos sobre titanio mediante varios procedimientos, pero la microestructura y la electroquímica de estos recubrimientos son, sin embargo, bastante diferentes. Por otra parte, J. Tang et al.64 informaron que películas de Au depositadas galvanostáticamente usando un tren de pulsos de crecimiento sobre Ti policristalino difieren significativamente de la estructura observada en películas preparadas por electrodeposición continua y evaporación al vacío.
RRO sobre los electrodos
El estudio de la RRO fue realizado por voltamperometría cíclica a velocidades bajas de barrido (10 mV/s) en un ambiente saturado en O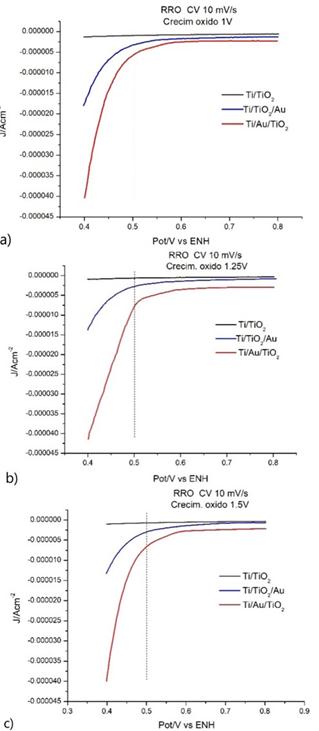 . En todos los casos la corriente catódica de la RRO fue normalizada por el área geométrica de la superficie para comparar la actividad catalítica de los electrodos, preparados bajo distintas condiciones en este trabajo. Una curva típica de polarización para la RRO se divide generalmente en tres regiones, a saber, región controlada cinéticamente, región mixta, controlada por cinética y difusión; y región controlada por difusión, a medida que decrece el potencial. La región controlada cinéticamente representa la parte donde la tasa de reducción de O
. En todos los casos la corriente catódica de la RRO fue normalizada por el área geométrica de la superficie para comparar la actividad catalítica de los electrodos, preparados bajo distintas condiciones en este trabajo. Una curva típica de polarización para la RRO se divide generalmente en tres regiones, a saber, región controlada cinéticamente, región mixta, controlada por cinética y difusión; y región controlada por difusión, a medida que decrece el potencial. La región controlada cinéticamente representa la parte donde la tasa de reducción de O es lenta con un pequeño incremento en el módulo de la densidad de corriente a potencial decreciente. Mientras que, en el área mixta controlada por la cinética y la difusión este incremento es mucho mayor. En la región controlada por difusión, la densidad de corriente está determinada por la velocidad a la que se produce la difusión de los reactivos. El análisis cuantitativo del catalizador en términos de su actividad se puede hacer a partir de los dos parámetros, esto es; el potencial de inicio
es lenta con un pequeño incremento en el módulo de la densidad de corriente a potencial decreciente. Mientras que, en el área mixta controlada por la cinética y la difusión este incremento es mucho mayor. En la región controlada por difusión, la densidad de corriente está determinada por la velocidad a la que se produce la difusión de los reactivos. El análisis cuantitativo del catalizador en términos de su actividad se puede hacer a partir de los dos parámetros, esto es; el potencial de inicio  (
(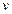 ) y la densidad de corriente a un potencial determinado,
) y la densidad de corriente a un potencial determinado, 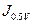 en nuestro caso. Cuanto mayor sea la densidad de corriente y más positivo sea el potencial de inicio, más activo será el catalizador hacia la RRO 65. Se analiza la región donde hay control cinético, es decir donde se inicia la RRO y la región próxima, esto es antes de que ocurra el control mixto. El valor de
en nuestro caso. Cuanto mayor sea la densidad de corriente y más positivo sea el potencial de inicio, más activo será el catalizador hacia la RRO 65. Se analiza la región donde hay control cinético, es decir donde se inicia la RRO y la región próxima, esto es antes de que ocurra el control mixto. El valor de  fue determinado por el potencial que separa la curva de la corriente catódica (saturada con O2) con la curva de corriente catódica (saturada con N2), con el fin de eliminar la corriente de carga de la doble capa eléctrica.
fue determinado por el potencial que separa la curva de la corriente catódica (saturada con O2) con la curva de corriente catódica (saturada con N2), con el fin de eliminar la corriente de carga de la doble capa eléctrica.
La Fig. 3 muestra las corrientes vs los potenciales de la región de estudio de la RRO para los tres casos (OEQ 1 V; 1.25 V y 1.5 V) para el TiO2 y los sistemas TiO2Ti/TiO2/Au y Ti/Au/TiO2.
Se puede ver, en todos los casos, de que el potencial de inicio de la RRO es siempre mayor para el sistema Ti/Au/TiO2, es decir: cuando se realiza la electrodeposición de Au antes de que se produzca el crecimiento electroquímico del óxido. Esto puede deberse a que hay más sitios con Ti , que es adonde reacciona el O
, que es adonde reacciona el O 9,55,66,67, mientras que, si el crecimiento del óxido se realiza antes, estos sitios son solapados parcialmente. La Tabla 3 muestra los valores de los potenciales de inicio () y de las corrientes (
9,55,66,67, mientras que, si el crecimiento del óxido se realiza antes, estos sitios son solapados parcialmente. La Tabla 3 muestra los valores de los potenciales de inicio () y de las corrientes ( ) al valor de 0.5 V de potencial (vs ENH).
) al valor de 0.5 V de potencial (vs ENH).
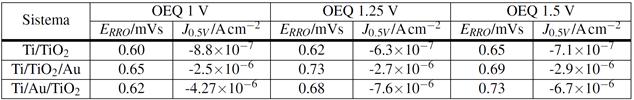
TABLA 3: Estudios electroquímicos de la RRO sobre los diferentes sistemas en 0.01M HClO
4
saturado con O
2
;=10 mVs-1 a 25ºC.
Se puede apreciar que los valores de corriente son mayores en el sistema Ti/Au/TiO2 en todos los casos. Con respecto a los potenciales de inicio , estos aumentan con el crecimiento de óxido en el sistema TiO2 y en el Ti/Au/TiO2. En el sistema Ti/TiO2/Au las corrientes, al valor de 0.5 V de potencial, son siempre superiores a las del sistema TiO2 e inferiores a las del sistema Ti/Au/TiO2.
Zejnilovic et al.36 estudiaron las propiedades catalíticas de la superficie de Ti recubierta con pequeñas cantidades de Au, Pt y Pd y encontraron que todos exhibieron una más alta actividad electroquímica, tanto para la reducción como para la evolución del oxígeno, que sus correspondientes superficies compactas, no obstante, el mejor rendimiento lo presentó el Pd, mientras que el rendimiento más pobre fue el del Au.
Si bien algunos autores como Mahé et al.68 y Gueneau de Muss y et al.69 trabajaron con sustratos de Ti, realizando anodización del mismo, seguido de la deposición de Pt; otros como L. Avalle et al.70 estudiaron el comportamiento del Ti, realizando primero la deposición del Pt seguida por el crecimiento potenciodinámico del óxido; pero no se hicieron estudios comparativos entre los procedimientos para el análisis de características comunes.
Por otra parte, F. A. Filippin71 estudió la RRO sobre electrodos de Ti con óxido crecido potenciodinámicamente en 0.01 M de HClO4, posterior a la electrodeposición de Pt (Ti/Pt/TiO2) y encontró para el sistema Ti/TiO2 un valor casi idéntico de que el apuntado en este trabajo para el mismo sistema.
IV. CONCLUSIÓN
En este trabajo hemos mostrado que, en la zona cinéticamente controlada, los sustratos con electrodeposición de Au sobre titanio antes del crecimiento potenciodinámico del óxido electroquímico (Ti/Au/TiO2) presentan una actividad electrocatalítica mayor para la RRO que aquellos sustratos a los cuales se les hace crecer el óxido electroquímico y luego se realiza la electrodeposición de Au (Ti/TiO2/Au).
Por otra parte, el hecho de que los depósitos de Au sobre los sustratos sin óxido electroquímico presenten menores valores de carga consumida, demuestra una mayor eficiencia en su preparación con respecto a los sustratos a los cuales se les hizo el depósito de Au posterior a la formación del óxido electroquímico.
Esto abre una clara posibilidad para la preparación de catalizadores más eficientes.

