











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkI. Introducción
El conocimiento sobre los efectos adversos que producen las emisiones de gases de efecto invernadero hacia la atmósfera - tales como el metano, los óxidos de nitrógeno y los óxidos de azufre - llevaron a un estudio más exhaustivo sobre las diferentes alternativas para captarlos antes de que sean expedidos por las chimeneas industriales, o bien, para directamente evitar su formación mediante la modificación en la relación de combustión en los motores presentes en las plantas que generan estos gases.
Por ejemplo, para llevar a cabo la captación de los gases contaminantes se utilizan lechos sólidos formados por combinaciones de metales de transición que actúan como filtros catalíticos, reteniendo mediante el proceso de adsorción los gases que llegan a su superficie e inclusive, disminuyendo la energía de activación necesaria para que entre ellos ocurran reacciones de oxidación y reducción.
El uso de óxidos de metales de transición como catalizadores ha conducido a avances económicos y tecnológicos muy importantes en los últimos años constituyéndose en una alternativa prometedora a los metales preciosos. La diversidad estructural de estos óxidos, así como su capacidad para ser mezclados, dopados y combinados con otros materiales como el grafeno, hacen de los óxidos metálicos de transición un tema altamente atractivo en la investigación, y supone el conocimiento de la interacción de diversas especies químicas con la superficie del catalizador, su estructura superficial y electrónica.1-5
En trabajos anteriores hemos realizado estudios sobre diversos óxidos, con énfasis en las reacciones de reducción de SO2 y oxidación de CH4 en presencia de oxígeno.6-11 Se ha llegado a la conclusión que el catalizador de óxido de cromo soportado en alúmina ( ) es el más eficiente para la adsorción de estos gases de efecto invernadero, debido a que el mismo no se desactiva por la presencia de gases ácidos, entre los cuales el más perjudicial para la actividad de un catalizador es el óxido de azufre (SO2) . Sobre esta base se continuó estudiando la interacción de SO2, CH4 y O2 sobre
) es el más eficiente para la adsorción de estos gases de efecto invernadero, debido a que el mismo no se desactiva por la presencia de gases ácidos, entre los cuales el más perjudicial para la actividad de un catalizador es el óxido de azufre (SO2) . Sobre esta base se continuó estudiando la interacción de SO2, CH4 y O2 sobre  . Hemos demostrado que la presencia de SO2 disminuye la energía de activación de la reacción de oxidación de metano.
. Hemos demostrado que la presencia de SO2 disminuye la energía de activación de la reacción de oxidación de metano.
En este trabajo presentamos un estudio teórico de la deshidrogenación de CH4 con O2 - en estado molecular y disociativo - previamente adsorbido en  (0001), mediante cálculos basados en la Teoría Funcional de la Densidad (DFT).
(0001), mediante cálculos basados en la Teoría Funcional de la Densidad (DFT).
A partir de los resultados proponemos algunas etapas del mecanismo de reacción y la existencia de intermediarios como el metanol, formaldehido y dioximetileno. Se establecen posibilidades interesantes para su exploración experimental.
II. Métodos
Los cálculos de energía total se realizaron basados en la Teoría Funcional de la Densidad añadiendo el parámetro U (DFT+U), a fin de investigar la adsorción individual y simultánea de moléculas de CH4 y O2 en la superficie  (0001). Para realizar estos cálculos se empleó el Paquete de Simulación Vienna Ab initio (VASP).12,13 La superficie de (0001) limpia se ha estudiado utilizando diferentes enfoques teóricos14-19 y existe un acuerdo general de que la superficie sufre fuertes relajaciones verticales. En este artículo tomamos en cuenta los efectos de correlación descritos por una repulsión de Coulomb in situ de tipo Hubbard, no incluida en una descripción funcional de densidad.9 Se seleccionó la cara (0001) porque en su estado natural, el
(0001). Para realizar estos cálculos se empleó el Paquete de Simulación Vienna Ab initio (VASP).12,13 La superficie de (0001) limpia se ha estudiado utilizando diferentes enfoques teóricos14-19 y existe un acuerdo general de que la superficie sufre fuertes relajaciones verticales. En este artículo tomamos en cuenta los efectos de correlación descritos por una repulsión de Coulomb in situ de tipo Hubbard, no incluida en una descripción funcional de densidad.9 Se seleccionó la cara (0001) porque en su estado natural, el  tiene este tipo de estructura en el 97.20% de su volumen, el cual se mantiene hasta temperaturas de aproximadamente 973 K. Esta característica fue verificada experimentalmente en etapas anteriores al presente estudio.7
tiene este tipo de estructura en el 97.20% de su volumen, el cual se mantiene hasta temperaturas de aproximadamente 973 K. Esta característica fue verificada experimentalmente en etapas anteriores al presente estudio.7
Las ecuaciones de Kohn-Sham se resolvieron utilizando el método de onda aumentada del proyector (PAW)8,13-15 y un conjunto de base de onda plana que incluye ondas planas de hasta 400 eV. Se utilizó el método DFT+U con valores J=1 y U=5.9 Los cálculos se realizaron utilizando pseudopotenciales basados en la aproximación de gradiente generalizada (GGA + U), en el esquema de Perdew, Burke y Ernzerhof.20-22 Para algunas propiedades estas aproximaciones dan mejores resultados que LDA utilizado en,11 en particular para geometrías moleculares y energías del estado fundamental.
Se logra alcanzar la convergencia de cada cálculo cuando las fuerzas sobre los iones son inferiores a 0.03 eV/Å. Las condiciones de contorno periódicas se aplican en las tres direcciones perpendiculares. La matriz de Hesse de segundas derivadas se determinó para las estructuras basales dentro de la aproximación armónica mediante diferencias finitas de dos lados, utilizando un paso de desplazamiento de 0.01 Å. Los átomos adsorbidos se desplazaron en los cálculos y la diagonalización de la matriz dinámica produce las frecuencias armónicas.
La superficie está modelada como una supercelda con base romboide, con una constante de red de 4.954 Å, con una altura de 20 Å, como se muestra en la Fig. 1. Cada capa de sustrato está compuesta por un átomo de cromo, tres átomos de oxígeno y un átomo de cromo, y tiene un espesor de 2.263 Å7.
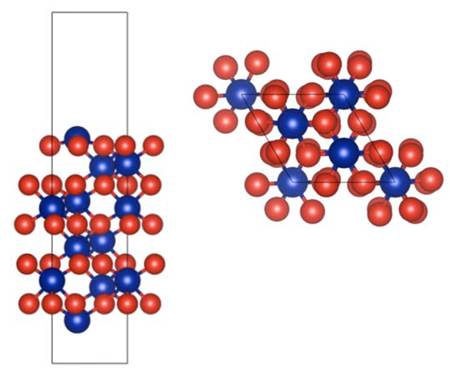
Fig. 1: Estructura de la superficie de (0001) limpia y optimizada. Constante de red: 4.954 Å, altura: 20 Å.
(0001) limpia y optimizada. Constante de red: 4.954 Å, altura: 20 Å.
La primera zona de Brillouin se determinó mediante una malla centrada en el punto gamma y sólo se utilizó un punto gamma para la supercelda cúbica empleada en la optimización de moléculas aisladas.13 La energía de adsorción de cada molécula de adsorbato se calcula como:
y sólo se utilizó un punto gamma para la supercelda cúbica empleada en la optimización de moléculas aisladas.13 La energía de adsorción de cada molécula de adsorbato se calcula como:
 (1)
(1)
El primer término es la energía de la configuración optimizada para la molécula de adsorbato relajada unida a la superficie limpia. El segundo término es la energía de la molécula de adsorbato en fase gaseosa optimizada (aislada) y el tercer término es la energía de la superficie optimizada. Según esta definición, los valores negativos de corresponden a configuraciones estables. La adsorción de oxígeno en estado molecular y disociado fue estudiada en.11 En estado molecular, el oxígeno se adsorbe con dos geometrías diferentes con aproximadamente la misma energía de adsorción. En una de éstas, ambos átomos de oxígeno se adsorben en el mismo átomo de Cr, mientras que en la otra, se une un solo oxígeno, como se muestra en la Fig. 2. En forma disociada, dos átomos de oxígeno se unen al mismo átomo de Cr y esta configuración es la más estable energéticamente como se muestra en la Fig. 3. Los valores de las energías se reportan en la Tabla 1.
corresponden a configuraciones estables. La adsorción de oxígeno en estado molecular y disociado fue estudiada en.11 En estado molecular, el oxígeno se adsorbe con dos geometrías diferentes con aproximadamente la misma energía de adsorción. En una de éstas, ambos átomos de oxígeno se adsorben en el mismo átomo de Cr, mientras que en la otra, se une un solo oxígeno, como se muestra en la Fig. 2. En forma disociada, dos átomos de oxígeno se unen al mismo átomo de Cr y esta configuración es la más estable energéticamente como se muestra en la Fig. 3. Los valores de las energías se reportan en la Tabla 1.

Fig. 2: Configuraciones más estables para la adsorción de O 2 en Cr 2 O 3 (0001) en estado molecular, con E ads = -1.48 eV (a,b) y E ads = -1.46 eV (c,d). VerTabla 1.
Fig. 3 Configuraciones más estables para la adsorción de O
2
en Cr
2
O
3
(0001) en estado disociativo, con
La adsorción de las especies CH4, CH3, CH2 y CH, se estudió tanto en la superficie limpia como en coadsorción de oxígeno, utilizando las geometrías optimizadas.
Se exploraron diferentes cuatro sitios de adsorción y diferentes orientaciones. Así, se obtuvieron las energías de adsorción de la configuración más estable para cada uno de los sistemas, junto con las longitudes de los enlaces, las variaciones en los ángulos formados entre los átomos de esas especies, y las nuevas especies formadas.
III. Resultados
No se hallaron configuraciones de adsorción estables del metano (CH4) sobre la superficie limpia, ni con oxígeno molecular preadsorbido en Cr2O3(0001). En presencia de oxígeno preadsorbido en forma disociada, se produce la disociación del metano y la formación de metanol con una energía de formación de , como se observa en la Fig. 4.

Fig. 4: Configuración más estable para la adsorción de CH4 en O2 disociativo sobre Cr2O3(0001): Sitio 3, con Eads 

El metilo (CH3) se adsorbe sobre la superficie limpia de Cr2O3 (0001), hallándose la mayor energía de adsorción en el sitio 3 (Fig. 5), con una energía de adsorción de  . En presencia de oxígeno preadsorbido en estado molecular sobre Cr2O3 (0001) el CH3 se adsorbe sin interactuar (Fig. 6), con una energía de adsorción de
. En presencia de oxígeno preadsorbido en estado molecular sobre Cr2O3 (0001) el CH3 se adsorbe sin interactuar (Fig. 6), con una energía de adsorción de 

Fig. 6: Configuración más estable para la adsorción de CH3 con O 2 molecular sobre Cr2O3 (0001): a y b). Sitio 2, con
En presencia de oxígeno preadsorbido en estado disociado el CH3 se disocia dando lugar a la formación de un OH que se une luego al carbono de alguna especie CHx. Las especies CH2 y el CH se adsorbieron en los cuatro sitios estudiados sobre la superficie limpia del sustrato (Figs. 7 - 8), con energías de adsorción de

Fig. 7: Configuraciones más estables para la adsorción de CH2 en Cr2O3 (0001), ambas con Eads = -3.53 eV: a y b). Sitio 1, c y d). Sitio 4.

El CH2interactúa con el oxígeno preadsorbido tanto en estado molecular como disociado dando lugar a la formación de formaldehído en estado gaseoso, con una energía de  (Fig. 9), y a dioximetileno adsorbido sobre Cr
2
O
3
(0001) con una
(Fig. 9), y a dioximetileno adsorbido sobre Cr
2
O
3
(0001) con una  (Fig. 10).
(Fig. 10).

Fig. 9: Configuración más estable para la adsorción de CH
2
en O
2
molecular en Cr
2
O
3
(0001): a y b). Sitio 2, con

Fig. 10: Configuración más estable para la adsorción de CH2 en O2 disociativo en Cr2O3 (0001): a y b). Sitio 3, con
Finalmente se estudió la adsorción de CH sobre oxígeno preadsorbido en estado molecular sobre Cr2O3 (0001), donde se observa la disociación del O2 y la formación de la especie H-C-O adsorbida como se muestra en la Fig. 11. La energía obtenida para la configuración más estable es de  . Sin embargo, no se halló una geometría estable para la misma especie sobre oxígeno preadsorbido en estado disociativo. Los resultados se resumen en la Tabla 2.
. Sin embargo, no se halló una geometría estable para la misma especie sobre oxígeno preadsorbido en estado disociativo. Los resultados se resumen en la Tabla 2.

Fig. 11: Configuración más estable para la adsorción de CH en O2 molecular sobe Cr2O3 (0001): a y b). Sitio 1, con 

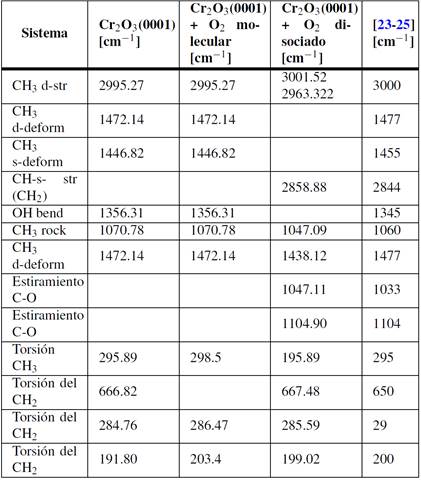
Los modos vibracionales para las especies CH x adsorbidas sobre la superficie limpia de Cr 2 O 3 (0001) se muestran en la Tabla 3 en comparación con datos de la bibliografía.
IV. Conclusiones
En este trabajo se realizaron cálculos DFT de la adsorción de las especies formadas por la deshidrogenación de CH 4 sobre la superficie limpia de Cr 2 O 3 (0001) y en presencia de oxígeno. Nos propusimos revelar posibles etapas de la reacción de oxidación de CH 4 sobre este catalizador, con el propósito de comparar con investigaciones experimentales en curso. El CH 4 es uno de los principales gases de efecto invernadero y puede ser retenido por adsorción antes que sea expedido por chimeneas industriales con lo que su emisión puede reducirse o eliminarse. El O 2 está presente en los gases de chimeneas, pero en ausencia de un catalizador adecuado no reacciona con CH 4 . El Cr 2 O 3 es un catalizador adecuado que conduce a la oxidación completa del CH 4 con formación de H 2 O y CO 2 .
Nuestros estudios muestran que el CH 4 no se adsorbe sobre la superficie limpia de Cr 2 O 3 (0001), ni en presencia de oxígeno adsorbido en estado molecular. En presencia de oxígeno adsorbido en estado disociado el metano pierde un átomo de hidrógeno vía la formación de un OH. El mismo mecanismo pudo verificarse para el CH 3 , que tampoco interactúa con el oxígeno adsorbido en estado molecular.
Asimismo, las especies CH3, CH2 y CH, pueden coexistir en la superficie ya que se adsorben en múltiples sitios diferentes. Por lo tanto, la oxidación del metano ocurre vía la formación de metanol, por interacción de OH y CH3. La formación verde de metanol a partir de la hidrogenación del CO 2 utilizando diversos catalizadores metálicos y bimetálicos soportados sobre alúmina fue propuesta en.26,27 El proceso requiere la captura de CO 2 y la producción verde de H 2 . Nuestra investigación señala hacia una alternativa adicional que merece ser explorada experimentalmente.
La interacción de CH 2 con O 2 molecular dió como resultado la formación de formaldehido en estado gaseoso. Se formó dioximetileno con una conformación estable en estado adsorbido por interacción de CH 2 con oxígeno disociativo, y la formación de H-C-O por interacción de CH sobre O 2 molecular, previa disociación del O 2 . Estas especies se encontraron experimentalmente en la oxidación de metanol sobre catalizadores de vanadio soportado sobre TiO.28 Medidas experimentales de la oxidación de metano sobre Cr 2 O 3 serán presentadas en un trabajo posterior.
Los modos vibracionales para especies adsorbidas sobre Cr 2 O 3 calculadas mediante DFT están en buena concordancia con los resultados de la bibliografía, lo que prueba la validez del DFT como forma de cálculo para energías de adsorción de gases sobre superficies de óxidos metálicos.

