










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de neurocirugía
versión On-line ISSN 1850-1532
Rev. argent. neurocir. v.21 n.1 Ciudad Autónoma de Buenos Aires ene./mar. 2007
La circulación cerebral en condiciones normales y patológicas. Parte I: hipertensión endocraneana e isquemia secundaria
Horacio Fontana, Héctor Belziti, Flavio Requejo, Mario Recchia, Sebastián Buratti
Hospital Central de San Isidro. Servicio de Neurocirugía.
RESUMEN
Las isquemias en pacientes que han fallecido por TEC grave se distribuyen predominantemente en territorios centrales y serían secundarias a distorsión y oclusión de los vasos perforantes por desplazamiento y hernia. Algunas lesiones centrales, podrían ser primarias.
La isquemia por disminución de la PP por hipertensión endocraneana es menos frecuente que la debida a distorsión vascular. Por lo tanto, pareciera ser más importante para la fisiopatología de la isquemia, el desplazamiento y hernia, que la presión de perfusión. El infarto se produce cuando el flujo no puede abastecer las necesidades metabólicas mínimas (penumbra) es decir, es menor de 20 ml/ 100gr/min, correspondiente a una PP entre 25 y 40mm Hg. No conocemos bien los factores que pueden prolongar la penumbra.
Es posible que en focos traumáticos la PP sea menor que la medida con las técnicas habituales, pero debería investigarse en qué casos produce isquemia. Por la lesión primaria, estas zonas pueden tener necesidades metabólicas despreciables.
La oxidación de la glucosa puede estar impedida en zonas isquémicas y acumularse metabolitos ácidos que son dañinos para la neurona. Es posible que las ondas en "plateau" no sean patógenas por sí mismas sino por causas estructurales concomitantes. Las oscilaciones no rítmicas de volumen son normales probablemente en todos los órganos. Oscilaciones de volumen de la lesión podrían ser otro factor desencadenante de ondas A.
La pulsación cerebral es transmitida a las venas intracraneanas y, a través de la columna sanguínea, a los senos durales, por las características de caja cerrada del cráneo.
La craniectomía descompresiva podría ser forzada por una terapia de la presión de perfusión exagerada. Presiones de perfusión por encima de 50 mm Hg podrían provocar edema y hemorragia en zonas contusas en vasoparálisis.
La craniectomía descompresiva está indicada en casos de edema de reperfusión, y debería realizarse precozmente. Las venas cerebrales no se colapsan durante el proceso de hipertensión endocraneana.
La sangre de los senos sigmoideos puede tener contenidos diferentes de uno u otro lado en casos patológicos, mientras que éstos son similares en pacientes normales.
Estudios del flujo o composición química de la sangre en el seno recto podrían ser de ayuda para la terapéutica y el pronóstico de pacientes con lesiones cerebrales agudas. Los cambios de velocidad circulatoria en los golfos yugulares podrían dar una idea aproximada de las variaciones de flujo cerebral global.
Palabras clave: Circulación cerebral; Presión de perfusión; Hipertensión endocraneana.
SUMMARY
The ischemic lesions in dead patients from severe head injury are distributed predominantly in central territories, and could be secondary to distortion and occlussion of perforant vessels during encephalic desplacement and herniation. Some central lesions could also be primary.
The ischemic lesions secondary to diminution of CPP during intracranial hipertension are less frequent than those due to vascular distortion. Thus, it seems physiopathologically more important desplacement and herniation than CPP.
A cerebral infarct occurs when CBF is inadequate to support the basic metabolic needs or is lower than 20 ml/ 100 gm/ min., corresponding with a CPP between 35 - 40 mm Hg. The while during which the cell rests viable is not well known.
Within traumatic foci, CPP could be lower than mesured by general methods, but ischemia sould not obligatorily follow it. The metabolic needs of those areas could be lowered because of primary cellular necrosis. The oxidative metabolism of glucose could be impaired during ischemia, producing accumulation of deleterious acid metabolites to the neuron.
It is possible that "plateau waves" be pathogenic not by itself, but by concomitant structural causes. Nonrhytmic oscilations of volume are normal probably in all organs. Oscillations in volume of the same lesion could be another cause of plateau waves. Decompressive craniectomy could be forced by an exaggerated CPP therapy. CPP over 50 mmHg could produce edema and hemorhage in contused brain in vaso paralysis.
Early decompressive craniectomy is indicated in cases with reperfusion edema. The cerebral veins does'nt collapse during intracranial hypertension. The composition of blood in one sigmoid sinus can be different from that of the other side in pathological cases, and is similar in normal subjects. Study of the flow or chemical composition of blood in the sinus rectus could be helpful for the prognosis and therapeutics in patients with acute cerebral disease.
Changes in flow velocity at the jugular bulbs coud give an approximation about global CBF changes.
Key words: Cerebral circulation; Perfusion pressure; Intracranial hypertension.
Actualmente, el concepto dominante para el tratamiento de los pacientes con hipertensión endocraneana es el de evitar la producción de lesiones secundarias por isquemia cerebral, debidas a la disminución de la presión de perfusión.
Esto ha llevado a dos tipos de línea para guiar el tratamiento, actuando cada una sobre uno de los factores que regulan el flujo sanguíneo cerebral en condiciones patológicas: la hipertensión endocraneana y la presión de perfusión.
Se han realizado incluso, estudios con un brazo de "mantenimiento del flujo sanguíneo cerebral" y otro de "tratamiento de la hipertensión endocraneana", sin haberse encontrado diferencias significativas en el resultado neurológico1.
Existen varios métodos para evaluar la perfusión cerebral como el Doppler transcraneano, la diferencia A-V de 02, la medición del flujo mediante el Doppler laser, etc.
Nuestro objetivo en esta revisión es intentar hacernos una idea lo más acabada posible de la circulación cerebral en condiciones normales y patológicas refiriéndonos no sólo pero preferentemente, al traumatismo craneoencefálico, para ayudarnos a conformar una base que nos permita interpretar en su justo valor, la información que nos aportan los mencionados métodos y los efectos de las terapéuticas en boga. Las citas serán las mínimas necesarias y las más básicas, para sostener nuestras tesis, ya que se trata de una revisión crítica.
1) Mecanismos de producción de la isquemia, según los datos de la anatomía patológica
Los mecanismos aceptados por los cuales se produce una isquemia cerebral en el neurotraumatizado son básicamente tres: 1) distorsión de pequeños vasos profundos, 2) compresión y distorsión de un gran vaso 3) disminución de la presión de perfusión.
a) Distorsión de pequeños vasos profundos. Los poco comprendidos fenómenos intracraneanos en el momento del traumatismo, pueden, como consecuencia de distorsiones vasculares bruscas, generadas en una escala de décimas o centésimas de segundo, generar daño vascular que se manifieste en una isquemia cerebral prácticamente inmediata o que se agrava en los instantes siguientes al traumatismo. Este tipo de daño vascular, había sido ya entrevisto por Norman Dott, quien pensaba que cuanto más brusca la distorsión vascular, más graves sus resultados2. Se refería, entre otros, a los delicados vasos hipotalámicos, que otros autores han encontrado que efectivamente se pueden afectar en los daños primarios cerebrales3. Es posible que4 también se puedan producir isquemias en el territorio de algunos vasos mesencefálicos en casos mortales de trauma craneano, aunque la isquemia como daño cerebral primario no está bien vista por la opinión neuropatológica predominante5.
En cambio, es ampliamente aceptado el daño de los vasos profundos secundario a desplazamiento y hernia encefálicos. Aquí los patólogos suelen referirse a las lesiones producidas en el mesencéfalo y protuberancia, por los desplazamientos horizontales y verticales que estas estructuras sufren en los distintos tipos de hernia. También muchos casos de daño de vasos hipotalámicos son atribuidos a esta causa con más benevolencia que a daño isquémico primario4,5.
Como vemos en la tabla 16, en dos grupos de pacientes con TEC grave, de los cuales la mayoría (87%) habían sufrido hipertensión endocraneana, un 81% presentaban isquemia del hipocampo, seguramente debida a un proceso de desplazamiento y hernia del mismo, concomitante al proceso de hipertensión endocraneana, como Hume Adams había demostrado previamente3.
Tabla 1: Lesiones isquémicas en dos grupos de pacientes fallecidos por TEC. Datos obtenidos de Graham et al 19896. Obsérvese el predominio de lesiones isquémicas centrales.
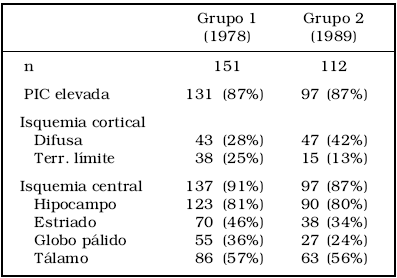
Pero el 57% de los pacientes fallecidos tenían isquemias en el tálamo, así que más de un 44% de los pacientes con hipertensión endocraneana presentaban lesiones isquémicas en este órgano. ¿Por qué no atribuir al menos algunas de estas isquemias a distorsión de los vasos perforantes o de sus vasos de origen, que irrigan el tálamo?
Las arterias comunicantes posteriores suelen estar involucradas en el proceso de hernia del uncus por ejemplo, y dan origen a las arterias t álamo perforantes anteriores o pedículo túbero- talámico de Foix e Hillemand, que pueden así verse comprometidas.
Lo mismo puede suceder con ramos profundos de la arteria cerebral posterior, tambi én frecuentemente involucrada en la hernia del hipocampo.
Seg ún estos datos podemos decir sin temor a equivocarnos, que al menos la mitad de los pacientes con isquemia hipocámpica (hipertensión endocraneana) presentaban una isquemia del tálamo, posiblemente debida a distorsión vascular en buena parte de ellos.
a) Distorsión de un gran vaso. Hemos hablado recién del ejemplo más típico, que es el infarto en el territorio de la arteria cerebral posterior por este mecanismo, durante la hernia transtentorial descendente.
b) Disminución de la presión de perfusión. Estas isquemias suelen afectar a la corteza cerebral y pueden ser difusas o de territorio límite.
Como vemos en la Tabla 1, son menos frecuentes que las isquemias del hipocampo (desplazamiento y hernia e hipertensión endocraneana) y que las del tálamo, igualando en cambio a las del cuerpo estriado. Es bien sabido que este núcleo sufre isquemia en cuadros de hipoperfusión cerebral, comportándose como una zona límite, especialmente el putamen y caudado7.
Así que es probable que isquemias profundas en este territorio, tengan que ser atribuidas al menos en parte, a disminución de la perfusión, como las corticales. Su clasificación junto a las talámicas en la mencionada tabla, confunde quizá un poco las cosas.
Lo que estos datos expresan es que la frecuencia de estas isquemias centrales entre los pacientes con hipertensión endocraneana fue inferior a las del tálamo, favoreciendo así la sospecha, que podrían estar relacionadas con mecanismos fisiopatológicos diferentes.
2) Aspectos clínicos de la lesión secundaria
a) Desplazamiento y hernia. Los fenómenos dinámicos intracraneanos producidos por una lesión expansiva de crecimiento rápido, como p.ej. un hematoma subdural agudo, generan desplazamiento y hernia encefálicos, con la consiguiente distorsión vascular central, producida en una escala de minutos a pocas horas.
Las isquemias así causadas tienen la característica de ser, bajo ciertas condiciones, reversibles. Esto es, si se pueden solucionar antes de que aparezca infarto o hemorragia en los territorios correspondientes. Pero debemos tener en cuenta que dan poco tiempo, por lo que la celeridad en su tratamiento o el tratamiento preventivo de lesiones que están mostrando alteraciones compatibles con el desplazamiento y hernia encefálicos antes de su descompensación8, es el desiderátum, porque una vez desencadenado el cuadro clínico de la hernia, los resultados pueden ser decepcionantes.
El predominio de lesiones de este tipo en la calota mesencefálica en pacientes con traumatismo de cráneo, hace que los cuadros no sólo pierdan rápidamente su reversibilidad, sino que sean, muchos de ellos, incompatibles con la vida9.
Marshall8 ha definido como lesión quirúrgica en estas condiciones, a aquella que tiene un modesto volumen por cierto, mayor de 25 cm3.
b) Lesión secundaria por hipertensión endocraneana.
La hipertensión endocraneana produce una disminución del flujo sanguíneo cerebral, por disminución de la presión de perfusión (PP).
Los estudios de isquemia en la autopsia de pacientes que han sufrido de hipertensión endocraneana pos traumatismo de cráneo, tienen la dificultad de no poder discernir en algunos casos, entre isquemia por lesión primaria, por lesión secundaria a desplazamiento y hernia, o las producidas por el descenso de la PP.
El predominio de lesiones en el tálamo y los núcleos de la base y el hipocampo6, como vimos, hace pensar que en muchos de estos casos, estas lesiones son debidas mas bien al desplazamiento y hernia o a la lesión primaria, que al descenso de la PP.
En cambio, los casos de isquemia cortical difusa o las lesiones de última pradera, serán seguramente debidos al descenso de la presión de perfusión, pero son minoría y, aunque no se discrimina en la publicación, muchas de ellas deben obligatoriamente coincidir con lesiones centrales (que son mayoría). Por otro lado, las elevaciones sostenidas de la presión intracraneana serían generadas en muchos casos, por los procesos de desplazamiento y hernia encefálicos10.
Según esta interpretación, estaríamos en condiciones de asegurar que en los pacientes fallecidos por un proceso de hipertensión endocraneana, predominan isquemias provocadas por distorsión vascular central, secundaria a desplazamiento y hernia, o eventualmente primaria. Estas lesiones no se evitan ni se previenen con una terapia de la PP.
Las lesiones por descenso de la PP serían en su mayoría, acompañantes de las anteriores.
Es posible que este sea el motivo por el cual la PP "baja" (<70 mmHg) no sea un predictor adecuado de mal pronóstico en algunos estudios11. Estos autores preconizan el tratamiento de la PIC, aún antes de tenerla monitoreada, guiándose por los datos estructurales como habíamos esbozado nosotros10.
En la figura 1 podemos observar un resumen esquemático de nuestra concepción. Obsérvese que las lesiones por distorsión o compresión vascular, se encuentran en la línea media, como es de esperar.

Fig. 1: Lesiones isquémicas en pacientes fallecidos por TEC, en base a datos de (3, 6, 13). A: Dibujo de la cara medial del cerebro tomado de Brodmann (12). 1) isquemia en el hipocampo. 2) lesiones isquémicas por compresión de grueso vaso (cerebral anterior y posterior) por la hernia debajo de la hoz y transtentorial descendente. Estas son las lesiones que caracterizan la anatomía patologica de pacientes que han fallecido con hipertensión endocraneana según Hume Adams13. B: corte de tipo Brissaud redibujado de (14). Se le han superpuesto en rojo las zonas con lesión isquémica por distorsión o compresión vascular. Obsérvese que son todas vecinas a la línea media en el tálamo o el hemisferio. En verde o azul: zonas con isquemia por hipoperfusión. En verde: isquemias de áreas de transición. En azul: isquemia difusa.
Hemos marcado con 1 la lesión del hipocampo, porque a nuestro juicio es debida en forma directa a la hernia, que a su vez, desencadena el resto del cuadro.
3) La circulación cerebral en condiciones patológicas
¿Qué sucede en los pacientes que no fallecen?
¿Puede el descenso de la PP por debajo de ciertos límites producir lesiones discapacitantes más allá de las generadas por la lesión primaria?
Muy probablemente sí, pero la interpretación fisiopatológica dista de ser simple.
Para que se produzca una lesión isquémica, el flujo sanguíneo cerebral debe estar por debajo de las necesidades metabólicas mínimas de la zona comprometida.
a) La "perfusión miserable". La penumbra isquémica es ese nivel de flujo sanguíneo que, aunque abolida la función, permite la supervivencia de la célula. Este nivel de flujo, para un tejido neural que trabaja normalmente, está alrededor de 20ml/100gr/min, lo que se consigue con una PP de aproximadamente 25 mm de Hg15.
Zonas del cerebro más o menos extensas que se hayan mantenido por encima de estos valores de PP o de flujo, durante episodios de riesgo, ya sea por aumentos descontrolados de la presión intracraneana o por hipotensión sistémica, o ambos, podrían aún haber escapado de sufrir una lesión isquémica definitiva.
Por otro lado, los pacientes que se encuentran en coma traumático tienen una disminución franca del metabolismo cerebral16,17, que podría hacer menos vulnerables a la isquemia a extensas áreas de tejido neural, haciendo más tolerables descensos de la PP por debajo de 40- 50 y por encima de 30 mm Hg.
Es posible pues, que aún en condiciones de mala perfusión cerebral, existan áreas en donde la isquemia es reversible por períodos importantes. Sería interesante, conocer esta variable temporal, y si hay factores que puedan prolongarla.
b) La regulación química. Aún en casos en donde la autorregulación esté agotada por descenso de la PP por debajo de 40-50mmHg, los vasos pueden mantener su reactividad a poderosos estímulos químicos generados en el área isquémica: pCO2, pH, adenosina. El aumento de estos sustratos produce una vasodilatación, que al reducir la velocidad de flujo sanguíneo capilar, permite una mayor extracción de O2 por parte del tejido cerebral.
Un aumento de la DAVO2, estará mostrando que estos mecanismos están activos (hipoperfusión compensada) 16.
c) La utilización de la glucosa. Asumimos que el cerebro se comporta como una máquina que extrae la energía para su funcionamiento de la oxidación completa de la glucosa a CO2 y H2O, y que es altamente eficiente en esta tarea.
La acumulación de metabolitos intermedios ácidos como piruvato-ácido láctico y su paso a la sangre venosa, ha sido interpretada como signo de isquemia severa16 y funcionamiento del tejido nervioso en anaerobiosis.
Durante la activación, el metabolismo de la glucosa por parte del tejido nervioso se haría en dos etapas, interviniendo en la primera (anaerobiosis) los astrocitos, que pasan los sustratos ácidos a la neurona, para que ésta desarrolle exclusivamente aquella parte del metabolismo de la glucosa que se hace en aerobiosis18.
Es posible que la detención del metabolismo neuronal por insuficiente aporte de oxígeno durante la penumbra, pueda generar un desacople metabólico entre el astrocito y la neurona (aparición de metabolitos ácidos), que podría sin embargo, ser reversible en algunos casos16.
d) La protección cerebral. Los métodos de protección cerebral contra la isquemia han consistido históricamente en tratar de disminuir el metabolismo neuronal a un mínimo, e históricamente también, han dado escaso resultado.
Si tenemos en cuenta lo comentado en el párrafo c), quizá pudiera ser útil además, una modulación de la función del astrocito. Esta modulación conseguiría evitar la acumulación de metabolitos ácidos perjudiciales19 tanto fuera como dentro de una neurona hipofuncionante.
La disminución del aporte de glucosa al cerebro hipometabólico, tanto endógena como exógena (aportada durante el tratamiento), como es de práctica en la actualidad, podría ser un paso inicial hacia esta modulación. Aparentemente durante la isquemia habría liberación masiva de glutamato20, que es el principal activador de la vía glucolítica en el astrocito18. Entre otros efectos, como toxicidad directa (excitotoxicidad) p.ej., podría también producirse el mencionado aumento de metabolitos ácidos, por este mecanismo, ya que las células gliales son más resistentes a la isquemia20.
4) "El cerebro es perfundido durante la sístole". (L. Marshall21, pág. 548).
Esta sorprendente aseveración acompañada de la autoridad de quien la formula, pero expresada sin apoyo bibliográfico explícito, nos hace pensar primero: ¿Qué sabemos acerca de las condiciones normales de la circulación cerebral? Y en seguida: ¿Será realmente así?
a) Volumen, presión y pulso. La contracción cardíaca durante la sístole tiene dos efectos: a) impulsa un volumen sanguíneo bruscamente al sistema arterial, con lo cual empuja hacia delante la sangre ya contenida en él, haciéndola progresar hacia el sistema capilar. b) al mismo tiempo, la distensión de la aorta, que es elástica, genera una onda de pulso, que se transmite por su pared. Esta onda se propaga mucho más rápido que el movimiento generado en la sangre22.
El pulso de las arterias, no significa pues, el paso de la sangre impulsada por la sístole, debajo de nuestros dedos, sino simplemente la transmisión por parte de la pared arterial de la onda del pulso.
Esta onda tiene sin embargo, una función importante: impulsar la sangre contenida en las arterias hacia delante durante la diástole, dándole una mayor continuidad a la circulación, que de lo contrario, sería solamente sistólica.
La presión generada por la contracción sistólica puede ser medida en cualquier arteria periférica, con pequeñas diferencias, a medida que nos alejamos del corazón. La elasticidad arterial hace que la presión no caiga a cero en las arterias durante la diástole, permitiendo la descarga de un volumen igual al impulsado por el corazón, hacia el sistema capilar y las venas. La resistencia periférica generada por el tono de las arteriolas de pequeño calibre y los capilares, hace que la pulsatilidad del movimiento de la sangre se atenúe hasta desaparecer, del lado venular del capilar.
b) Cambios de volumen de un órgano durante el ciclo cardíaco: el pletismograma. La variación de volumen producida durante el ciclo cardíaco, por la entrada de sangre en cualquier órgano, ha podido ser medida por los fisiólogos, mediante el pletismógrafo22-24 (Fig. 2). Se trata de una caja inextensible, llena de agua, que transmite hacia un delgado tubo de vidrio, las variaciones de volumen del órgano encerrado en él, que se traducen en variaciones del nivel líquido en el tubo.

Fig.2. Pletismograma de dedo, tomado de (25). A: respiración .Pl: pletismograma. R: pulso radial. Obsérvese el retardo de la onda respecto al pulso- La flecha roja muestra la aparición de una onda parecida a P2 de la figura 3 durante la espiración.
"Para facilitar la colocación del órgano en su interior, los pletismógrafos se abren como si se tratara de dos valvas unidas por una bisagra. Los bordes cierran ajustadamente en toda su extensión menos en una parte que, debido a sendas escotaduras, queda un orificio por donde pasará el pedículo vásculo nervioso del órgano. Las variaciones de presión de la cámara formada entre el órgano y el pletismógrafo, actuando sobre la cápsula inscriptora, permiten obtener el trazado de las variaciones de volumen del órgano o pletismograma" 22.
El cr áneo es, en cierta medida, el pletismógrafo del cerebro.
Las pulsaciones que acostumbramos ver durante la cirugía, pueden ser transformadas en una oscilación de la columna líquida de un manómetro de Ayer, o en variaciones rápidas de la onda de presión intracraneana, con cada ciclo cardíaco.
La onda de presión es entonces, en cierta medida, un pletismograma del cerebro (fig. 3).
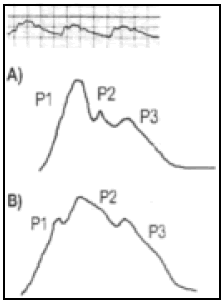
Fig. 3. Onda de PIC tomada de (26). Arriba: registro habitual. A: registro con PIC normal. Obsérvese el parecido con el pletismograma de dedo de la figura 2. B: registro con PIC alta, la onda P2 se acentúa.
Efectivamente, el cerebro pulsa con cada sístole, como cualquier otro órgano, pero esto no quiere decir que no haya circulación durante la diástole.
c) La circulación pulsante. La pulsación de las venas cerebrales y de los senos durales.
Aaslid et al han encontrado que, contrariamente a lo que se podría suponer, el flujo en el seno recto de la duramadre es pulsátil en el 100% de los casos en sujetos normales27 (Fig. 4).
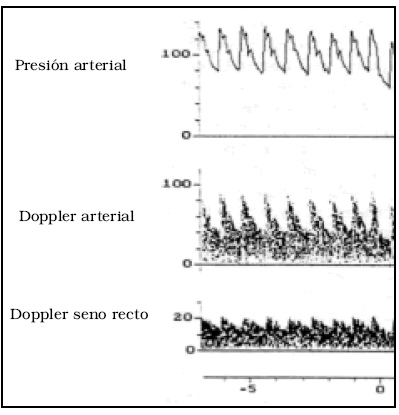
Fig. 4. Trazados de presión arterial, registro Doppler de las arterias de la base y del seno recto, tomado de (27) que muestra la pulsatilidad del flujo en éste, y también la continuidad de la circulación en diástole. La línea roja muestra la simultaneidad de los picos de presión y flujo, lo cual demuestra que el pico del seno es transmitido por el pulso cerebral.
Creo que por esta razón, estos autores concluyen que "la circulación cerebral normal es un lecho vascular de alto flujo, relativamente rígido, y de baja resistencia periférica".
Quiz á para ellos, esta rigidez vascular, permitiría que los cambios de presión en el sistema arterial, sean transmitidos a las venas, a través del sistema de baja resistencia, lo que explicaría la pulsatilidad del flujo en los senos.
Sin embargo, la explicaci ón de esta pulsatilidad podría ser muy otra: "Si se introduce una cánula a través del occipital en la prensa de Herófilo, se ve pulsar la sangre venosa sincrónicamente, pero en sentido opuesto con la pulsación arterial"23.
Para Starling, los cambios rápidos de volumen cerebral son transmitidos a las venas en el cráneo cerrado, a través de sus delgadas paredes por eso pulsa la sangre venosa de los senos. En este tipo de registros, ¡el sistema venoso actuaría como el líquido de transmisión del pletismógrafo!
Tanto el mencionado trabajo de Aaslid y col. como las curvas de presión, son importantes para nosotros, porque aunque la circulación es pulsátil aún en los senos venosos, estos registros muestran que es continua durante todo el ciclo cardíaco. Coincidentemente, las curvas de Doppler de la carótida interna muestran una velocidad elevada de flujo incluso en momentos terminales de la diástole, que alcanzan al 30- 40% de la velocidad del pico sistólico28.
As í que, en condiciones normales, la sangre circula por el cerebro tanto en sístole como en diástole, mal que le pese a Marshall.
Con cada latido card íaco ingresan al cráneo unos 10-12 cm3 de sangre, de los cuales, posiblemente la mitad lo hace durante la sístole. Este pequeño volumen es sin embargo, suficiente para generar el "pulso cerebral".
d) El pulso venoso en la papila. El efecto de "caja inextensible" y la transformación del flujo continuo de las venas en pulsátil por este motivo, es también evidente en el fondo de ojo. Estamos tan acostumbrados a ver el "pulso venoso" en el fondo de ojo normal, que pocas veces nos hemos preguntado acerca de su origen. El ojo, como el cráneo, es un órgano inextensible, por lo que la sangre de la vena central de la retina es "exprimida" hacia fuera con cada sístole29,30.
La dependencia del drenaje venoso de la retina de la PIC, a pesar de ser conocida desde hace tiempo, ha sido actualizada recientemente31.
e) La presión en los capilares cerebrales. Es posible que la presión de los capilares cerebrales sea variable durante el ciclo cardíaco32, debido a que el aumento de PIC que se produce con cada latido, aumenta la presión tisular, y por lo tanto, tendría que transmitirse también a los capilares. Así, los capilares pulsarían con el pulso cerebral, pero disminuyendo su calibre durante la sístole, comenzando el "efecto pletismógrafo" tan evidente en las venas.
f) La resistencia vascular cerebral. Estimo que la aseveración de que el lecho vascular cerebral es "de baja resistencia", no es tan exacta.
De este modo, pues, hay que decir que el ventrículo derecho fue creado por causa de los pulmones, y para la traslación de la sangre y no para su nutrición. Además nos parecería inconveniente admitir que los pulmones tienen necesidad de alimento tan abundante, suministrado con tan grande impulso y tan puro y cargado de espíritus (como el que sale inmediatamente del corazón), como sucede con el cerebro que exige mejor alimento por la sustancia particularmente pura del cerebro admirable y divina... (William Harvey. De motu cordis33).
Sabemos que el pulmón es el único órgano de la economía por el que circula todo el gasto cardíaco. Podemos pues suponer sin temor a equivocarnos, que es el órgano con menor resistencia vascular periférica, y así lo demuestran las características de presión de su sistema.
Por el cerebro, circula sólo el 15% del gasto cardíaco, a una presión cinco veces mayor que en el pulmón, por lo que calculamos que su resistencia periférica es al menos veinte veces mayor que la de este órgano*. Esta resistencia está distribuida de la siguiente manera: 17% en los grandes vasos hasta el polígono de Willis, 26% desde el polígono hasta los vasos de 200 micrones, 32% hasta los capilares, 25% los capilares y venas, en un paciente normotenso34.
La vascularización de diferentes áreas cerebrales puede aumentar notoriamente según los requerimientos metabólicos. Así, durante una crisis epiléptica, la circulación puede incrementarse hasta un 200%15.
Tomando esta circunstancia como un máximo, debemos aceptar que la resistencia vascular cerebral es al menos dos veces mayor que la que se podría alcanzar en condiciones de requerimiento metabólico extremo. ¿Y en condiciones patológicas?
d) El flujo sanguíneo en condiciones patológicas. Durante la hipertensión endocraneana la PP desciende, y se pone en juego el mecanismo de autorregulación, que produce una vasodilatación de las arteriolas de resistencia, para mantener el flujo.
Estudios teóricos35, experimentales36 y clínicos37, con ecografía Doppler transcraneana han demostrado que en estas condiciones, y a medida que la PIC aumenta, las velocidades sistólicas de la sangre en las arterias de la base del cerebro aumentan levemente o se mantienen, mientras que las velocidades diastólicas van disminuyendo, hasta que con PICs de 35 a 55mm Hg en condiciones experimentales en gatos y una PP de 9 a 47 mmHg, la circulación en diástole cesa.
Como aproximación, se podría decir que la circulación en diástole tenderá a cesar, cuando la presión diastólica intracraneana se acerque a la presión diastólica sistémica38. Recordemos que en nuestros registros de PIC nos referimos habitualmente a la presión intracraneana media.
Experimentalmente, el electrocorticograma comenzaría a alterarse mientras la circulación diastólica disminuye, se agravaría seriamente cuando sólo hay flujo sistólico y desaparecería cuando el flujo sistólico está disminuyendo o cesa36.
El flujo tisular medido con láser Doppler, sin embargo, parecería no modificarse hasta que la PP alcanza valores de 35mm Hg y el índice de pulsatilidad es de 2 (los valores del láser Doppler podrían ser equívocos en estas circunstancias)39.
Analizando estos datos, vemos que el cese de la circulación diastólica, se produce aproximadamente en el límite inferior de la autorregulación, o algo por debajo de ella, es decir, en condiciones extremadamente críticas de la circulación cerebral. Estas serían quizá, las condiciones en que "el cerebro se perfunde en sístole". ¿Podrían mantener una actividad circulatoria mínima, intermitente, que prolongue la penumbra, o una función celular mínima? - Posiblemente, quizá, por poco tiempo.
De estos datos parece surgir que, contrariamente a la afirmación de Marshall, la circulación en diástole es un componente esencial de la perfusión del cerebro normal. La función del tejido nervioso se mantendrá normal, mientras la presión diastólica sea efectiva. A pesar del llamado de atención de Hassler38, hace casi 20 años, ha preocupado poco este aspecto de la circulación cerebral cuando se trata de condiciones patológicas. Hasta donde conocemos, no existen estudios detallados de la circulación en diástole durante períodos de hipertensión endocraneana, y de haberlos, no han sido tomados debidamente en cuenta para el manejo de esta patología.
La mejor información acerca del flujo cerebral durante la diástole la brindarían la PP diastólica y el índice de pulsatilidad en el Doppler transcraneano. El monitoreo neurofisiológico (EEG especialmente) podría brindar información valiosa complementaria.
5) La pendiente hidrodinámica
a) El "colapso" de las venas puente. Por la ley de Poiseuille, la presión y la velocidad del flujo irán disminuyendo progresivamente a lo largo de un tubo de calibre constante, sometido a un flujo constante, dependiendo básicamente de la longitud del tubo y de su calibre, si otras variables, como la viscosidad del fluido, se mantienen constantes, como se puede ver en el esquema tomado de Starling23 (Fig. 5).
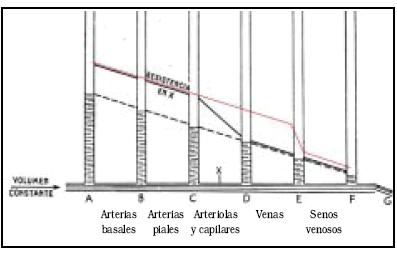
Fig. 5. Caida de la presión en un sistema de tubos con volumen de flujo constante, modificado de Starling23. En la estrictura X, la presión aumenta por detrás de la misma, y la velocidad de flujo aumenta a su nivel (línea continua). La línea roja muestra la teórica distribución de presiones en un caso de hipertensión endocraneana con PP en el límite inferior de la auto regulación, según nuestra opinión.
La interposición de una resistencia a determinado nivel, aumentará mucho la presión por detrás y no la cambiará por delante, para que el flujo se mantenga constante. El gradiente de presión es el responsable del paso del líquido de uno a otro segmento, y la velocidad deberá aumentar a nivel de la estrictura, para mantener el flujo. Si ahora trasladamos esta simple experiencia a la circulación cerebral en condiciones de hipertensión endocraneana, recordaremos que para mantener el flujo, la resistencia vascular cerebral disminuye por vasodilatación arteriolar (auto-regulación). Esto permite mantener el flujo contra una presión de perfusión en disminución. La presión en los capilares y las venas aumenta, por transmisión más directa de la presión arterial a estos sectores.
Dec ía Starling en la descripción de la "resistencia artificial" (resistor) de su preparado cardiopulmonar, conectada a la aorta40 (Fig.6).

Fig. 6. Detalle de la resistencia periférica artificial del preparado cardiopulmonar de Starling40. Ver descripción en el texto.
"Éste consiste en un tubo de goma delgada [R], colocado dentro de otro más amplio de vidrio [herméticamente cerrado][T]. Mediante una jeringa, se puede bombear aire en el tubo de vidrio [W]… Es evidente que si la presión afuera es mayor que la interior al tubo de goma, éste se colapsa y si algún flujo ha de existir a través del tubo de goma, la presión del líquido debe ser mayor que la exterior."
Lo mismo puede aplicarse al sistema vascular cerebral. Mientras el flujo se mantenga constante, la presi ón en los capilares y las venas deberá ser necesariamente un poco mayor que la PIC. Esta es la principal razón para decir con seguridad, que las venas cerebrales no se colapsan durante períodos de hipertensión endocraneana con auto-regulación presente.
Nakagawa et al 41 pretenderían haber demostrado lo contrario en una experiencia muy bien conducida en perros, pero en donde estos autores parecen haber interpretado desacertadamente sus datos experimentales, que, analizados detenidamente, de ninguna manera argumentan en favor de un colapso venoso durante los ensayos de hipertensión endocraneana.
Estudios ulteriores, han demostrado fehacientemente el hecho de que las venas no se colapsan42 y sólo disminuyen su calibre levemente (Fig. 7) durante la hipertensión endocraneana.

Fig. 7. Variaciones del diámetro de una vena puente con distintas presiones intracraneanas, levemente modificado de (42). SLS: seno longitudinal superior. Se puede constatar que las venas no se colapsan incluso con presiones intracraneanas muy altas. Solamente disminuyen su calibre levemente.
b) La circulación en los senos durales. Como los senos durales son incompresibles, igual que los orificios por donde desembocan en ellos las venas cerebrales, la PIC no se transmite a ellos, o lo hace en forma muy imperfecta a través de la columna sanguínea pulsátil de las venas. Debe haber entonces un gradiente importante entre la presión de las venas cerebrales (algo mayor que la PIC) y la de los senos durales (cercana a cero), durante la hipertensión endocraneana, que ha sido bien documentada por Nakagawa et al41 (Fig. 8).

Fig. 8. Variación de la presión al mover un catéter desde una vena puente hacia el SLS, durante un experimento de hipertensión endocraneana. Tomado de (41). Obsérvese que la presión en las vena puente es algo mayor que la PIC, y que cuando el catéter entra en el SLS la presión cae prácticamente a cero. Al final del trazado, se observa la curva del pulso cerebral en ambos trazados (pletismograma). El trazado del SLS NO PULSA porque se está midiendo la presión estática (lateral) del mismo.
Como el flujo se mantiene constante mientras la autorregulación esté preservada, debe haber un aumento muy grande de la velocidad de la sangre a nivel de los orificios de entrada a los senos como pediría la lógica del simple experimento de Starling (Fig. 5).
Es decir que, en condiciones de hipertensi ón endocraneana, el gradiente de presión endovascular se iría desplazando, en buena parte, del sector arteriolo- capilar al de los orificios de los senos venosos, a medida que la auto-regulación va siendo exigida por los aumentos de PIC. Este efecto será máximo, en momentos previos a la vasoparálisis (Fig. 8).
Como el flujo se mantiene constante, la velocidad de la sangre en los senos durales no debería aumentar, pero sí es probable que lo haga su pulsatilidad, que podría ser medida con una sonda Doppler en un agujero sobre la tórcula por ejemplo.
c) la hipertensión en los capilares. Mencionamos más arriba, que al vasodilatarse las arteriolas, durante los descensos de PP por hipertensión endocraneana, la presión arterial es transmitida más directamente a los capilares, aumentando por lo tanto, la presión endoluminal de los mismos.
Por el juego de presiones de la ley de Starling, al aumentar la presión endoluminal, debería salir líquido de los capilares hacia el intersticio, produciéndose edema. Esto no sucede, sin embargo, porque hay un aumento de la presión extraluminal (PIC = presión tisular) que no sólo equilibra a la intraluminal, sino que fue la causa desencadenante del proceso.
Durante la hipertensión endocraneana las zonas no afectadas por la lesión primaria y con auto-regulación preservada, podrán verse congestivas, pero no edematosas.
6) Las ondas "plateau"
Estas ondas de la presión endocraneana fueron descriptas por Lundberg en su monografía43. Aparecen en pacientes con hipertensión endocraneana, y son aumentos de la misma de amplitud variable, de instalación y finalización rápida y de duración larga, medible en minutos, entre 5 y 10 habitualmente. Son las ondas A, y no las B como reza un conocido libro de neurointensivismo44.
Han intrigado a los investigadores desde el comienzo. Lundberg las atribuyó a episodios de vasodilatación de explicación oscura.
Más recientemente, Rosner les atribuyó un origen más lógico. Él opina que estas ondas se producen durante la hipertensión endocraneana, cuando la presión de perfusión baja por alguna razón, desencadenándose una respuesta auto-reguladora, que sería la causante de la vasodilatación, que en un cráneo con "compliance" disminuida, genera el "plateau"45.
Estas ondas están rodeadas de una aureola de patogenicidad, que ya le había sido atribuida por Lundberg, aunque por sus mismos datos habría quizá, que revisar este concepto:
"Un aumento en la presión del líquido ventricular (VFP) hasta 60 mm Hg o menos, raramente estuvo acompañada por síntomas salvo cefalea o inquietud, y a menudo ocurrió sin síntoma ninguno.
Ondas entre 80 y 100 mm Hg estaban como regla, acompañadas de síntomas marcados y no raramente por trastornos de conciencia. Ondas de este orden, sin embargo, también ocurrieron, a veces, sin signos de disfunción cerebral. En las relativamente pocas ocasiones en que la presión excedió 100mm de Hg, síntomas de algún tipo u otro, fueron observados, usualmente estupor o coma. Presiones sobre 100 mm de Hg sin trastorno demostrable de conciencia, fueron registradas ocasionalmente en un caso"43.
Once años después escribía: "No hay una relación consistente entre la altura de las ondas de presión y la severidad de los síntomas acompañantes. Grandes ondas (>80 mm Hg) pueden ocurrir sin síntomas o con sólo síntomas menores, mientras elevaciones moderadas, bajo ciertas condiciones, pueden estar acompañadas de síntomas alarmantes, incluso paro respiratorio. Las observaciones clínicas indican que es posible que síntomas severos ocurran en pacientes con lesiones infratentoriales con circulación sistémica en fallo. Es también posible que el estado real de la autorregulación y el grado de estrés en el tronco cerebral por herniación/distorsión sean factores de significación46".
Es decir, hay que buscar otra causa para explicar por qué algunos pacientes con ondas plateau tienen síntomas de hernia y otros no. La causa aparente, pareciera ser, según sus dichos, que el paciente está efectivamente herniado o tiene una lesión descompensada en la fosa posterior.
Así que sería útil revisar hasta dónde se extiende realmente el poder patogénico de estos fenómenos de la PIC.
Lo notable es que Lundberg relata valores de PIC durante estas ondas, no permisibles en la actualidad, y sin embargo muchos pacientes de su serie no presentaron síntomas durante las mismas.
En realidad, las variaciones de volumen de un órgano son normales, y ya fueron descriptas por Götz en 1935, en su pletismógrafo de dedo, como aumentos duraderos y no rítmicos del volumen25, de duración de varios segundos a pocos minutos, y de amplitud variable.
Estas variaciones están en el orden de una a dos centésimas del volumen del órgano, así que trasladadas al cerebro, significarían un aumento del volumen de unos 10-20 cm3.
Si existieran, ¿por qué no se manifestarían estas variaciones en individuos con presión intracraneana normal? Porque en estado normal el cráneo no es un pletismógrafo perfecto, ya que es capaz de compensar la variación lenta de volumen mediante la salida de LCR.
Cuando aparece el bloqueo a la circulación de LCR, cualquiera sea su causa, y la hipertensión endocraneana consecuente10, el cráneo se transforma en rígido (baja compliance), y cumple con las condiciones para un buen pletismógrafo. En estas condiciones, un aumento en 10-20 cm3 del contenido intracraneano, sería más que suficiente para producir un "plateau".
Habría que aceptar por un lado, que ambos mecanismos patogénicos pueden ser válidos, y por otro, que las ondas "plateau" significarían en principio, que la autoregulación está preservada en grandes áreas del cerebro. Un mecanismo que no parece explorado por Lundberg ni otros autores que se han referido al tema, son las variaciones de volumen de la lesión misma.
Los cambios en la composición química de la sangre y de la PP, generan cambios en el volumen y la velocidad circulatoria cerebral, que pueden ser transmitidos a los vasos tumorales, cuya reactividad posiblemente sea menor o nula, actuando en realidad como una vasoparálisis.
Esto podría generar oscilaciones en el volumen tumoral, cuyos vasos actuarán pasivamente ante estos cambios. La masa puede también agrandarse efectivamente, como en el caso de los hematomas, generando aumentos de la presión que serán mal manejados por un sistema cuya "compliance" está agotada o casi, por el bloqueo a la circulación de LCR generador del aumento de PIC.
Hay mucha confusión en la literatura respecto a los tres tipos de ondas descriptas por Lundberg en pacientes con hipertensión endocraneana, a saber: ondas A o en plateau que acabamos de discutir, ondas B y ondas C. Un respetable libro de neurointensivismo dice en su capítulo 2 (básico):… "(las ondas A) deben distinguirse de las ondas B del pulso y ondas C de la respiración"44. Nunca asoció Lundberg las ondas B al pulso ni las C a la respiración. No deseamos aquí, discutir este tema en profundidad, pues sale de nuestro objetivo. Más bien invitamos al lector, si no lo ha hecho, a leer la hermosa monografía de Lundberg, para que pueda constatar cuánto se tergiversa su aporte por nuestros días.
7) La vasoparálisis
Este término designaría un estado de los vasos de resistencia, en que no responden a cambios en la presión de perfusión, porque se encuentran fuera del rango en que esta respuesta es posible, ni tampoco, al estímulo de la pCO2.
Es un estado de dilatación máxima del lecho arteriolo- capilar.
Estos reflejos son muy sensibles a diferentes noxas, por lo que en la HSA o en lesiones traumáticas, sobre todo en las contusiones, pueden existir zonas amplias del parénquima con vasoparálisis.
¿Cómo es posible la circulación en estas condiciones?
En realidad, el flujo cerebral puede estar severamente comprometido y los autores coinciden en que depende pasivamente de la presión de perfusión.
Cuando la PP es alta, el flujo podría ser incluso mayor que las necesidades metabólicas de la zona afectada, generándose una hiperemia.
Como la presión a nivel de los capilares no puede ser regulada, dependerá también pasivamente de la presión de perfusión. Si la presión de perfusión supera en más de 50-60 mmHg a la presión tisular (PIC), existiría aquí sí, la posibilidad de que salga líquido hacia el intersticio, generando edema.
Es más, como los capilares pueden estar también dañados, tenderán a ser más permeables a las proteínas del plasma, especialmente la albúmina, disminuyendo la presión oncótica dentro del vaso, para aumentar en el intersticio.
A mayor presión de perfusión mayor edema, y en casos extremos, incluso hemorragia.
Así, las zonas contusas presentarán frecuentemente un cuadro de congestión (hiperemia) y edema o hemorragia, que se agravarán con PP > de 50 mmHg. Estas lesiones evolutivas pueden llevar a la necesidad de la evacuación de coágulos y resección de la zona contusa, o a la necesidad de una craniectomía descompresiva, si la lesión es más difusa.
Es posible que uno de los factores que han llevado a que esta operación se haya puesto en boga en los últimos años, sea la moda de una terapia agresiva con la PP (>70 mmHg). Habría que ver si una terapia más prudente con la TA y los líquidos ayuda a disminuir la cantidad de amputaciones craneanas que significa esta operación.
Hay que tener en cuenta también, que "el umbral para el breakthrough puede haberse desplazado a niveles más bajos de presión arterial después de trauma craneano grave"47.
También es posible que éstos sean los factores que hacen que trabajos recientes48 encuentren beneficiosa una PP entre 50 y 60 mm Hg, pero siempre menor de 60.
Para Rosner, en 1995, la presión de perfusión debía estar por encima de 70 mmHg49.
Para Marshall en el 2000, por encima de 60. Pero más de 70 podría ser perjudicial21.
Para Elf et al en el 2005, la PP debería estar por encima de 50 mm Hg, pero nunca por encima de 6048.
Quizá nos hemos estado acercando a una prudencia beneficiosa con el correr de la década.
8) El "breakthrough".
No sabemos cuánto influye esta situación en el cuadro total de pacientes con TEC o HSA, pero podría no ser despreciable, como acotábamos más arriba. Se ha demostrado experimentalmente que, en condiciones de hipercapnia (vasodilatación máxima) con hipertensión arterial por encima del límite de la autoregulación (estado parecido a la vasoparálisis), durante el breakthrough, se produce hipertensión endocraneana, que es secundaria a la hipertensión venosa y que llega a haber hipertensión del seno longitudinal superior50.
La extravasación de líquido y proteína se produce predominantemente a nivel vénulo- capilar.
La sobrecarga venosa producida por el breakthrough, dificulta el drenaje adecuado de zonas vecinas cuyo avenamiento converge con las afectadas, produciendo congestión en aquellas.
La administración de hipertensores puede producir estos fenómenos en zonas en breakthrough. El aumento de flujo cerebral en estos casos, producirá seguramente un aumento de la velocidad de la sangre en los senos durales, que se traducirá en un aumento de la presión hidrodinámica en los mismos51 en condiciones experimentales como las de Auer et al50 y menos probablemente en la clínica.
La medición continua de la velocidad del flujo en los senos durales mediante DTC, puede ser de ayuda para el control de las medidas terapéuticas, especialmente el aporte de líquidos y los hipertensores.
9) La reperfusión de zonas isquémicas. Sabemos que zonas cerebrales que han estado privadas de circulación pueden ser reperfundidas, si el tiempo de isquemia no es muy prolongado (ventana). Después de la reperfusión, pueden observarse dos fenómenos patológicos52:
a) hipoperfusión crónica. El tejido no se deja perfundir, y hay una disminución marcada del flujo en la zona, que no alcanza a más del 20% del original.
b) Fenómeno de "no reflujo". Hay daño de los capilares que se edematizan y dificultan el flujo sanguíneo "en parches", restableciéndose el mismo en otras áreas.
Aumentando la presión de perfusión, la recirculación puede aumentar, pero las lesiones capilares descriptas, permiten el pasaje de líquido y eventualmente sangre al intersticio, generándose un tipo de edema cerebral de muy difícil manejo médico.
Áreas de isquemia por oclusión de un vaso grueso de causa habitualmente embólica, o compresiones corticales extensas y brutales como se generan durante la formación de algunos hematomas subdurales agudos, producen este tipo de fenómeno con cierta frecuencia.
El mejor tratamiento para este edema es la craniectomía descompresiva52, que se puede hacer preventivamente al operar un hematoma subdural agudo, y que se debería programar precozmente en las oclusiones silvianas reperfundidas espontáneamente o por trombolisis.
El fenómeno de no reflujo se presenta luego de una isquemia global superior a los 10 minutos53 y parcial (un vaso) de hasta unas seis horas52. Aunque por la ventana temporal el cuadro sea totalmente reversible, parece demostrado experimentalmente, que después de la reperfusión hay un período inicial, de hiperemia, con hipertensión endocraneana y EEG isoeléctrico, de unas dos horas, seguido por otro de hipoperfusión de unas ocho horas53, debido probablemente a edema capilar. Después de la isquemia, la mayor dificultad para la revascularización parece estar pues, a nivel del lecho capilar.
10) El drenaje venoso cerebral
Nos queremos referir aquí a la anatomía de la confluencia de los senos y las consecuencias que se derivarían de la misma.
Según los anatomistas clásicos, habría tres tipos de confluencia de los senos54:
a) tipo I: los dos senos laterales son de igual tamaño, el seno longitudinal divide sus aguas en forma equivalente hacia ambos lados, y el seno recto termina en el centro de la confluencia se forma así, lo que lamamos "prensa de Herófilo" (20%).
b) Tipo II: los senos laterales son asimétricos, habitualmente el derecho es más grande y recibe al seno longitudinal superior. El izquierdo es menor, y recibe al seno recto, o viceversa (28%).
c) Tanto el seno longitudinal como el recto, dan dos ramas, una para cada seno lateral (34%). Pero habitualmente predomina una de ellas, pareciéndose entonces al tipo anterior.
Es decir, que en aproximadamente 1/3 de los casos, no hay mezcla de las sangres venosas en los golfos yugulares, recibiendo uno (casi siempre el izquierdo) la sangre de las venas profundas y el otro la de las venas superficiales de ambos hemisferios55. En otro grupo de pacientes, difícil de definir numéricamente, la situación sería parecida. En el resto, la sangre de ambos lados tendrá una mezcla adecuada de los sistemas superficial y profundo (1/5 de los casos).
Estos hechos no son trascendentes en sujetos normales, porque es difícil que, en estas condiciones, haya diferencias significativas entre ambos lados como aseveraban Gibbs y col. hace años (56).
La situación sería diferente en condiciones patológicas, como han encontrado Metz y col. (57). En estos casos, midiendo en forma bilateral, hallaron diferencias significativas entre un golfo y otro, tanto en saturación de oxígeno como en contenido de lactato. Estas diferencias serían impredecibles en muchos casos para estos autores, por lo que el método puede calificarse de poco fiable.
Si nuestra concepción del daño predominante de los vasos profundos expuesta al principio es cierta, deberían ser más importantes a los efectos diagnósticos, los sucesos ocurridos en la sangre del seno recto, que drena el sistema de los vasos perforantes, sean estos mecánicos: velocidad de flujo, pulsatilidad, o metabólicos: DAVO2, contenido de lactato, etc.
Además, el desplazamiento horizontal y hernia encefálicos propios de los procesos de hipertensión endocraneana, pueden actuar directamente sobre las venas cerebrales internas y basales de Rosenthal, generando una disminución del flujo en el seno recto.
Las diferencias de composición de la sangre de los golfos yugulares en casos patológicos, podrían en buena parte ser explicadas por el drenaje variable del seno recto en la mayoría de los pacientes, como se describió más arriba.
Las posibilidades de derivación del flujo venoso por anastomosis con las venas superficiales o a través del parénquima cerebral hacia el sistema superficial, por las venas medulares anastomóticas descriptas por Schlessinger o de las venas trans cerebrales descriptas por Kaplan58, puede evitar quizá infartos venosos en el territorio del sistema profundo en estas circunstancias.
Aaslid et al27 han descripto una forma de insonar el seno recto en individuos sanos, que sin embargo, parece difícil de trasladar a los pacientes. Quizá sea una buena veta investigar en este sentido.
Es tentadora la idea de estudiar en forma sistemática el flujo en los golfos yugulares en forma continua, ya que reciben el producto del flujo cerebral, y variaciones de éste deberían manifestarse en ellos. Lamentablemente, son compresibles59, dificultando así los cálculos de flujo a partir de la velocidad de la sangre. Además, parte variable del flujo cerebral podría ser derivada por los plexos venosos raquídeos60.
DISCUSIÓN
Todo lo aquí expresado, es el resultado de un trabajo que, si bien basado en hechos sostenidos por datos de la bibliografía, es en muchos aspectos, altamente especulativo, por lo que algunas de sus aseveraciones deben ser consideradas como hipotéticas, y requerirían comprobación experimental o clínica.
Es incluso posible, que puedan ser refutadas simplemente a la luz de un conocimiento más amplio de una materia que creemos que, en general, se nos escapa a muchos neurocirujanos, o está en el límite de nuestro conocimiento.
Justamente, la revisión que hemos encarado, tiene la intención de situarnos en estos límites para preguntarnos qué sabemos de la circulación cerebral, y tratar de ampliarlos, para comprender mejor los sucesos fisiopatológicos a los que están sometidos quienes sufren un padecimiento cerebral agudo.
Los jóvenes, que inician su actividad como neurocirujanos o neurointensivistas, reciben una pequeña parte de esta información encadenada en forma aparentemente coherente, y referida a una lógica terapéutica de consecuencia inmediata. Lamentablemente, la realidad es más compleja, y el tamizado crítico de lo que se lee y aconseja en "guidelines" es necesario para preservar nuestro pensamiento y proteger a nuestros pacientes. Para ello, se precisa una base de conocimiento más amplia que la que habitualmente se nos suministra. Esperamos que este artículo contribuya a crear inquietud sobre estos temas especialmente entre sus lectores jóvenes (de edad o de espíritu).
Cito a Robertson1: "El enfoque de tratar a todos los pacientes manteniendo la PPC a un nivel alto para compensar la incapacidad del cerebro dañado para autorregular a niveles de PPC normal, puede ser muy simplista".
Es justamente por esto, que aprovecharemos para discutir en cierta profundidad, solamente la terapia de la PP.
A) El manejo de la PPC
En el curso evolutivo de un TEC grave la relación entre TAM y PIC puede presentar diversas alternativas, que son encuadrables entre algunas de las siguientes.
A) Hipotensión sistémica con PIC normal.
Este cuadro es más común en pacientes con daño neurológico de grado variable y con otras lesiones no neurológicas, con pérdida de volumen sanguíneo al exterior o con hemorragias internas. En general el paciente agrava sus signos neurológicos o trastorno del estado de conciencia, para recuperarlos casi siempre (según su daño primario y duración de la hipotensión), en forma parcial o completa, una vez solucionada la hipotensión con reposición de volumen. La latencia de la recuperación es variable, pero suele ser medible en minutos u horas y está posiblemente, en relación con la duración de la hipotensión.
El empeoramiento de los síntomas durante la hipotensión, es probablemente multifactorial, influyendo la disminución de la PPC, pero también la hipoxia tisular por la anemia frecuentemente asociada. Las zonas de daño primario, presentan autorregulación defectuosa o abolida y allí pueden producirse lesiones isquémicas secundarias por hipotensión, que agraven definitivamente el cuadro neurológico.
La sangre es la forma más adecuada de reposición de volumen en estos casos, con el agregado de coloides.
B) Normotensión sistémica con PIC normal
En este caso, estando compensado hemodinámicamente el paciente con TEC grave, se puede asumir que su sintomatología es enteramente debida a la lesión primaria. La intervención médica, está dirigida a mantener al paciente dentro de estos parámetros.
Es de hacer notar, que en estos casos, salvo insuficiencia respiratoria por lesión torácica o por daño neurológico primario, no estaría indicado el manejo con respirador del paciente, limitándose la intervención neurointensiva al cuidado adecuado de la vía aérea, prevención de aspiraciones, aspiración de secreciones, mantenimiento de la humedad y temperatura de las mucosas, drenaje postural y kinesioterapia respiratoria, etc.
C) Hipotensión sistémica con hipertensión endocraneana Esta es una asociación nefasta, que aunque conocida hace mucho tiempo61, no había recibido en general, una adecuada atención hasta principios de los 90.
Además de la arriba referida hipovolemia, hay una serie de situaciones en que el paciente con hipertensión endocraneana, corre el riesgo de hipotensarse:
a) Comienzo de la hiperventilación. Esta maniobra, especialmente si se utiliza PEEP, puede producir una disminución del retorno venoso y una eventual disminución del gasto cardíaco, que podrían llevar a la hipotensión arterial, habitualmente transitoria, hasta que la rémora aumenta la presión en las venas, restituyéndose el ingreso adecuado de sangre al tórax.
b) Drogas hipotensoras. Entre ellas, los depresores del SNC, que se usan para inducir la anestesia, o intubar y adaptar al paciente al respirador, o iniciar un coma barbitúrico terapéutico.
Si el paciente tiene PIC de 25-30 mmHg, un descenso moderado de la TAM a 65-70 mmHg por ejemplo, puede hacer entrar al paciente en la zona crítica de la autorregulación, de 40 mmHg de PPC, por debajo de la cual, puede aparecer isquemia cerebral difusa, que si no se corrige, puede llevar al paciente al daño cerebral severo, desencadenar un proceso de deterioro rostrocaudal o producir la muerte encefálica.
c) La inducción de la anestesia es un momento especialmente crítico para el paciente neuroquirúrgico agudo y el anestesiólogo debe estar siempre prevenido en este sentido.
Notemos que todas estas circunstancias son iatrogénicas y de ocurrencia común en un sector neurointensivo.
El protocolo propuesto por Rosner49 nos ha prevenido contra ellas, ha tenido eco, y en este aspecto es bienvenido.
Estas circunstancias deben ser adecuadamente previstas y evitadas, restringiendo el uso de la intubación y depresión del paciente y su ulterior respiración mecánica, a aquellos casos de indicación precisa.
Es posible que la mayor parte del éxito de la terapia de manejo de la PPC se deba a la prevención y tratamiento solícito de este tipo de situaciones.
D) Normotensión sistémica con hipertensión endocraneana
Estos casos son habituales en la T. intensiva. Si la PPC está por encima de 50 mm Hg, no hay necesidad de intervención terapéutica y ésta debe dirigirse al descenso moderado de la PIC. En pacientes no desplazados, el drenaje de LCR es un buen método, no así en aquellos que tienen desplazamiento1,10. Estos pueden ser manejados con manitol, hiperventilación moderada, y eventualmente, cirugía.
En este grupo de pacientes, especialmente aquellos con desplazamiento, los resultados son malos, cualquiera sea la modalidad terapéutica. Rosner y col. publican una mortalidad de 47% en pacientes que requirieron drogas hipertensoras para mantener la PPC49.
E) Hipertensión sistémica con hipertensión endocraneana
Si el paciente no es un hipertenso previo, se trata de una respuesta de CUSHING y es indicio de gravedad extrema. Si el problema no tiene solución quirúrgica, es poco probable la recuperación del paciente por medios médicos.
Resumen conceptual del manejo de la PP para el tratamiento de pacientes con riesgo de daño isquémico secundario.
Los intentos por mejorar la PP cerebral en pacientes con diversas patologías pueden ser beneficiosos, cuando van dirigidos a tratar o prevenir la hipotensión arterial sistémica. Este aspecto de la terapia de la PP debe ser especialmente enfatizado.
En cambio, su efecto es dudoso y puede ser perjudicial, cuando se trata a pacientes normotensos, cuya PP baja por aumento de la PIC. Es más, para Robertson et al1, "el protocolo dirigido al flujo sanguíneo cerebral, más que proteger de una PIC elevada, puede predisponer a una grave hipertensión endocraneana refractaria... Estudios experimentales han demostrado un aumento del edema vasogénico como consecuencia de la inducción de hipertensión y administración de líquidos".
Lo que surge, es que algunas de las terapias utilizadas actualmente, podrían en ciertos casos ser tan riesgosas, o aún más, que los mismos padecimientos tratados, confirmando aquello de "a grandes males, grandes remedios".
Como los otros temas fueron discutidos en cada uno de los ítems expuestos, los resumiremos en forma de conclusiones aproximadamente firmes en algunos casos o que deberían promover una investigación más exhaustiva en otros.
CONCLUSIÓN
1. Las isquemias encontradas en pacientes que han fallecido por TEC grave se distribuyen predominantemente en territorios centrales y fisiopatológicamente, serían secundarias a distorsión y oclusión de los vasos perforantes por desplazamiento y hernia. Algunas de las lesiones centrales, podrían ser primarias. Las isquemias debidas a disminución de PP por hipertensión endocraneana (difusas corticales) y/o hipotensión arterial (territorios límite cortical y/o central), son menos frecuentes que las debidas a distorsión.
2. Por lo tanto, pareciera ser más importante para la fisiopatología de la isquemia, el desplazamiento y hernia, que la presión de perfusión. La PP (<70 mmHg) no ha sido un buen predictor de resultado en diversos estudios.
3. La lesión cerebral por isquemia (infarto) se produce cuando el flujo no alcanza a abastecer las necesidades metabólicas mínimas es decir, es menor de 20 ml/ 100gr/min, correspondiente a una presión de perfusión entre 25 y 40 mmHg. Hay factores temporales (duración de la penumbra) que no conocemos bien y su conocimiento debería ser profundizado.
4. Algunos autores creen que en los focos traumáticos la presión de perfusión puede ser menor que la medida con las técnicas habituales62.
Es muy posible, pero debería investigarse en qué casos produce isquemia.
Muchas de estas zonas pueden presentar necrosis celular masiva como lesión primaria, y tener por eso, necesidades metabólicas despreciables.
5. La hiperglucemia puede ser un mecanismo fisiológico para hacer llegar este metabolito a zonas hipoperfundidas pero la utilización de la glucosa puede estar alterada en estas zonas y acumularse metabolitos ácidos que son dañinos para la neurona.
6.- Las ondas en plateau ó A de Lundberg, fueron vistas por este autor como patógenas. Sin embargo, en sus mismos escritos se describen pacientes con presiones intracraneanas no permisibles en nuestra era, que no presentaban síntomas.
Es posible que éstos se deban más bien a causas estructurales concomitantes, como el mismo autor refiere.
Las oscilaciones no rítmicas de volumen son normales probablemente en todos los órganos. Sería interesante saber si son sincrónicas.
Oscilaciones de volumen de la misma lesión podrían ser otro factor desencadenante de ondas A.
7. Todos los órganos pulsan. La pulsación cerebral es transmitida a las venas intracraneanas y, a través de la columna sanguínea, a los senos durales, por las características de caja cerrada del cráneo.
El flujo en los senos no sería pulsátil entonces, por transmisión del empuje sistólico de la sangre a través de la red capilar.
8. La craniectomía descompresiva podría ser forzada por una terapia de la presión de perfusión exagerada.
Es probable que presiones de perfusión por encima de 50- 60 mmHg puedan provocar edema y hemorragia en zonas contusas en vasoparálisis.
9. La craniectomía descompresiva está indicada en casos de edema de reperfusión, y debería realizarse precozmente.
10. Las venas cerebrales no se colapsan durante el proceso de hipertensión endocraneana, probablemente hasta la detención final del flujo sanguíneo cerebral.
11. La sangre de los senos sigmoideos puede tener contenidos diferentes de uno u otro lado en casos patológicos, mientras que éstos son similares en pacientes normales.
12. Sospechamos que la diferencia en casos patológicos puede estar dada por el desagüe del seno recto en uno u otro de los senos laterales, ya que trae la sangre de las zonas profundas del cerebro, sometidas a isquemias por distorsiones vasculares en los procesos de desplazamiento y hernia cerebral, antes de la disminución del flujo por descenso de la PP.
Las venas que en él desaguan también sufren seguramente, distorsiones obstructivas durante estos procesos.
13. El estudio del flujo o composición química de la sangre en el seno recto podría ser de gran ayuda para la terapéutica y el pronóstico de pacientes con lesiones cerebrales agudas.
14. Como los senos durales recogen el producto del flujo sanguíneo cerebral, los cambios de velocidad circulatoria en los golfos yugulares podrían dar una idea más aproximada de las variaciones de flujo global, que otros métodos.
Lamentablemente, los senos sigmoideos son compresibles probablemente en forma parcial. Este aspecto del seno sigmoideo merece un estudio especial.
El drenaje sanguíneo hacia los plexos vertebrales también complica esta táctica.
Nota
* Según la conocida fórmula: R = P/Q, que indica que la resistencia (R) es directamente proporcional al gradiente de presión (P) e inversamente proporcional al flujo (Q); tenemos para el pulmón R = 25mm Hg/ 5000cm3= 0,005 mmHg/cm3. Para el cerebro R = 90 mmHg/850cm3= 0,1058. La proporción 0,1058/0,005= 21,16, nos da la relación entre la resistencia cerebro vascular y la pulmonar.
1. Robertson CS, Valadka AB, Hannay HJ, y col. Prevention of secondary ischemic insults alter severe head injury. Crit Care Med 1999, 27: 2086- 95. [ Links ]
2. Dott NM. Brain, movement and time. Brit Med J 1960; XXX 12- 6. [ Links ]
3. Hume Adams J. The neuropathology of head injuries. Handbook of clinical neurology. Vinken, PJ, Bruyn, GW. Vol 23, Amsterdam. Nord-Holand, publ. Co., 1975. [ Links ]
4. Rosenblum WI, Greenberg RP, Seelig JM, Becker DP. Midbrain lesions: frequent and significant prognostic feature in closed head injury. Neurosurgery, 1981; 9: 613- 20. [ Links ]
5. McCormick WF. Pathology of closed head injury. En: Wilkins RH, Rengachary SS (Eds.): Neurosurgery, Vol. II, McGraw- Hill, New York, 1996. [ Links ]
6. Graham D, Ford I, Hume Adams J, Doyle D, Teasdeale G, Lawrence A, McLellan D. Ischaemic brain damage is still common in fatal non missile head injury. J Neurol Neurosurg Psych 1989; 52: 346-50. [ Links ]
7. Zülch KJ. The cerebral infarct. Pathology, patogénesis and computed tomography. Springer Verlag, Berlin, 1985. [ Links ]
8. Marshall LF, Bowers Marshall S., Klauber MR, van Berkum Clark, M y col. A new classification of head injury based in computerized tomography. J Neurosurg, 1991; 75: S14- S20. [ Links ]
9. Hume Adams J, Graham DI, Jennett B. The neuropathology of the vegetative state after an acute brain insult. Brain, 2000; 123: 1327- 38. [ Links ]
10. Fontana H, Belziti H. Hipertensión endocraneana y traumatismo encéfalo craneano. Revisión conceptual. Rev Arg Neurocir, 1995; 9: 73-83. [ Links ]
11. Juul N, Morris GF, Marshall SB, The Executive Comité of the Internacional selfotel trial, Marshall, LF. Intracranial hypertension and cerebral perfusion pressure: influence on neurological deterioration and outcome in severe head injury. J Neurosurg, 2000; 92:1- 6. [ Links ]
12. Brodmann K. Vergleichende Lokalisatinslehre der Grosshirnrinde in ihren Prinzipien dargestellt auf Grund des Zellenbaues. Leipzig: Barth, 1909. [ Links ]
13. Adams JH, Graham DI. The relatioship between ventricular fluid pressure and the neuropathology of rised intracraneal pressure. En: Brock, M, Dietz, H, eds: Intracranial pressure. Springer, Berlin, pp 250- 253. [ Links ]
14. Kretschmann HJ, Weinrich W. Neuroanatomía y tomografía computadorizada cerebral. Doyma, Barcelona, 1988. [ Links ]
15. Astrup J. Cerebrovascular physiology. En: Carter, LP, Spetzler, RF, Eds.: Neurovascular surgery. McGraw- Hill, Inc., New York, 1994. [ Links ]
16. Robertson CS, Narayan RK, Gokaslan ZL, Pahwa R, Grossman RG, Caram P Jr., Allen E. Cerebral arteriovenous oxygen difference as an estimate of cerebral blood flow in comatose patients. J Neurosurg, 1989, 70: 222- 30. [ Links ]
17. Sokoloff L. Neurophysiology and neurochemistry of coma. Exp Biol Med 1971; 4: 15- 33. [ Links ]
18. Magistretti PJ. Brain energy metabolism. En: Squire LR, Bloom FE, Mc Connell SK, Roberts JL, Spitzer NC, Zigmond MJ (eds.): Fundamental Neuroscience. Academic Press, Amsterdam, 2003. [ Links ]
19. Rovlias A, Kotsou S. The influence of hyperglycemia on neurological outcome in patients with severe head injury. Neurosurgery, 2000; 46: 335- 43. [ Links ]
20. Choi DW. The excitotoxic concept. En: Welch KMA, Caplan LR, REIS DJ, Siesjö BK, Weir B. (eds.): Primer on cerebrovascular deseases. Academic Press. San Diego, 1997. [ Links ]
21. Marshall LF. Head injury: recent past, present and future. Neurosurgery, 2000; 47: 546- 61. [ Links ]
22. Houssay BA. Fisiología humana. El ateneo. Buenos Aires, 1957. [ Links ]
23. Starling EH, por Lovatt Evans Ch. Principios de fisiología humana. Aguilar, Madrid, 1955. [ Links ]
24. Ganong WF. Manual de fisiología médica. El Manual Moderno, México, 1978. [ Links ]
25. Götz RH. Der Fingerplethysmograph als Mittel zur Untersuchung der Regulationsmechanismen in peripheren Gefässgebieten. Pflügers Archiv f d ges Physiol. 1935; 235: 271-87. [ Links ]
26. Citerio G, Andrews PJD. Intracraneal pressure. Part two: clinical applications and technology. Intensive Care Med, 2004; 30: 1882- 5. [ Links ]
27. Aaslid R, Newell DW, Stoos R, Sorteberg W, Lindegaard KF. Assessment of cerebral autorregulation dynamics from simultaneous arterial and venous transcranial Doppler recordings in humans. Stroke, 1991; 22: 1148-54. [ Links ]
28. Kerber CW, Liepsch D. Flow dynamics for radiologists. II. Practical considerations in the live human. AJNR, 1994; 15: 1076-86. [ Links ]
29. Baurmann G. Über die Entstehung und klinische Bedeutung des Netzhautvenenpulses. Dtsch Ophtalmol Ges 1925; 45: 53-9. [ Links ]
30. Reis W. Der Netzhautvenenpuls und der intrakranielle Druck. Ztschr. Augenheilk 1932; 78: 47-74. [ Links ]
31. Firsching R, Schütze M, Motschmann M, Behrens-Baumann W. Venous ophtalmodynamometry: a noninvasive method for assessment of intracranial pressure. J Neurosurg 2000; 93: 33-6. [ Links ]
32. Heistad DD, Kontos HA. Cerebral circulation. Handbook of Physiology, "The cardiovascular system III", pp 137-82, 1983. [ Links ]
33. Harvey W. De motu cordis. Estudio anatómico del movimiento del corazón y de la sangre en los animales. EUDEBA, Buenos Aires, 1970. [ Links ]
34. Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JLJr. Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Physiol, 1978; 234: H371-83. [ Links ]
35. Ursino M, Giulioni M, Lodi CA. Relationships among cerebral perfusion pressure, autoregulation, and transcranial Doppler waveform: a modeling study. J Neurosurg, 1998; 89: 255-66. [ Links ]
36. Nagai H, Moritake K, Takaya M. Correlation between transcranial Doppler ultrasonography and regional cerebral blood flow in experimental intracranial hypertension. Stroke, 1997; 28: 603-8. [ Links ]
37. Chan K-H, Miller JD, Dearden NM, Andrews PJD, Midgley S. The effect of changes in cerebral perfusion pressure upon middle cerebral artery blood flow velocity and jugular bulb cenous oxygen saturation after severe brain injury. J Neurosurg, 1992; 77: 55- 61. [ Links ]
38. Hassler W, Steinmetz H, Gawlowski J. Transcranial Doppler ultrasonography in raised intracranial pressure and in intracranial circulatory arrest. J Neurosurg, 1988; 68: 745-51. [ Links ]
39. Ungersböck K, Tenckhoff D, Heimann A, Wagner W, Kempski OS. Transcranial Doppler and cortical microcirculation at increased intracranial pressure and during the Cushing response: an experimental study on rabbits. Neurosurgery, 1995; 36: 147-57. [ Links ]
40. Starling EH. The Linacre lecture on "The law of the heart". Longmans, Green and Co., London, 1915. [ Links ]
41. Nakagawa Y, Tsuru M, Yada K. Site and mechanism for compression of the venous system during experimental intracraneal hipertensión. J Neurosurg, 1974; 41: 427-34. [ Links ]
42. Auer LM, Ishiyama N, Hodde KC, Kleinert R, Pucher R. Effect of intracranial pressure on bridging veins in rats. J Neurosurg, 1987; 67: 263-8. [ Links ]
43. Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psych Neurol Scand, 1960; 149(supp), p 138. [ Links ]
44. Ropper AH y col. Neurological and neurosurgical intensive care. Lippincott, Williams & Wilkins, Philadelphia, 2004, p. 23. [ Links ]
45. Rosner MJ, Becker DP. Origin and evolution of plateau waves. Experimental observations and a theoretical model. J Neurosurg, 1984; 60: 312-24. [ Links ]
46. Lundberg, N., Kjallquist, A., Kullberg, G., Pontén, U. y Sundbarg, G. Non-operative management of intracranial hypertension. Adv. and. Tech. Stand. Neurosurg. Springer, Wien, 1: 3-59, 1974. [ Links ]
47. Bouma GJ, Muizelaar JP, Bandoh K, Marmarou, A. Blood pressure and intracranial pressure-volume dynamics in severe head injury: relationship with cerebral blood flow. J Neurosurg, 1992; 77: 15- 9. [ Links ]
48. Elf K, Nilsson P, Ronne-Engström E, Howells T, Enblad P. Cerebral perfusion pressure between 50 and 60 mm Hg may be beneficial in head-injured patients : a computerized secondary insult monitoring study. Neurosurgery, 2005; 56: 962-71. [ Links ]
49. Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg,1995;83:949-62. [ Links ]
50. Auer L, Johansson B, MacKenzie ET. Cerebral venous pressure during actively induced hypertension and hypercapnia in cats. Stroke, 1980; 11: 180-3. [ Links ]
51. Parisi M. Temas de biofísica. Dos Santos, Buenos Aires, 1997. [ Links ]
52. Hossmann K-A. Hemodynamics of postischemic reperfusion of the brain. En: Weinstein PR y Faden AI (eds.): Protection of the brain from ischemia. Williams & Wilkins, Baltimore, 1990. [ Links ]
53. Melgar MA, Rafols J, Gloss D, Díaz FG. Postischemic reperfusion: ultrastructural blood- brain barrier and hemodynamic correlative changes in an awake model of transient forebrain ischemia. Neurosurgery, 2005; 56: 571-81. [ Links ]
54. Testut L, Latarget A. Tratado de Anatomía Humana. Tomo III. 9ª Ed. Salvat, Barcelona, 1973. [ Links ]
55. Sokoloff L. Anatomy of cerebral circulation. En: Welch, Caplan, Reis, Siesjö, Weir (eds.): Primer on cerebrovascular deseases. Academic Press, San Diego, 1997. [ Links ]
56. Gibbs EL, Lennox WG, Gibbs FA. Bilateral internal jugular Blood. Comparison of A-V differences, oxigen-dextrose ratios and respiratory quotiens. Am J Psychyat, 1945/1946; 102: 184-90. [ Links ]
57. Metz C, Holzschuh M, Bein T, Woertgen C, Rothoerl R, Kallenbach Taeger, K, Brawanski A. Monitoring of cerebral oxygen metabolism in the jugular bulb: reliability of unilateral measurements in severe head injury. J Cer Blood Flow Met, 1998; 18: 332-43. [ Links ]
58. Salamon G, Huang YP. Deep cerebral veins. En: Radiologic anatomy of the brain. Cap. 4. Springer, Berlin, 1976. [ Links ]
59. Krayenbühl HA, Yasargil MG. Cerebral angiography. Butterworths, London, 1968. [ Links ]
60. Eckenhoff JE. The physiologic significance of the vertebral venous plexus. SGO, 1970; 131: 72-8. [ Links ]
61. Christensen, JC. Comunicación personal.
62. Teasdale, G, en Comentario al trabajo (39).

