










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de neurocirugía
versión On-line ISSN 1850-1532
Rev. argent. neurocir. v.23 n.4 Ciudad Autónoma de Buenos Aires oct./dic. 2009
La circulación cerebral en condiciones normales y patológicas VI. El caso del vasoespasmo
Horacio Fontana, Héctor Belziti, Sebastián Buratt
Hospital Central de San Isidro. Servicio de Neurocirugía
Correspondencia: hojofontana@yahoo.com.ar
Recibido: septiembre de 2009.
Aceptado: octubre 2009
RESUMEN
En la base del cerebro tenemos un sistema arterial de alta velocidad y de gran disipación de energía de la masa circulante por rozamiento (flujo alterado), con vasos colaterales que se benefician de estos fenómenos, con una baja presión, que podría tener importancia en la patología isquémica a este nivel; y una zona distal, a la que la corriente sanguínea llega con menor velocidad y presión, comparativamente con los vasos extracraneanos del mismo calibre.
Es posible que el aumento de velocidad en las arterias de la base después de HSA se deba a una disminución de la resistencia periférica, por Suna abolición de la autorregulación. El aumento de velocidad disminuye la presión lateral en las arterias por el principio de la conservación de la energía, lo que a su vez produce disminución pasiva del diámetro de la arteria. Esto hace que el espesor de la pared aumente, aumentando el tono vascular, que disminuye el calibre del vaso, por lo que aumenta la velocidad, generándose un círculo vicioso. Otro factor que puede influir es la presión extravascular (PIC) y los coágulos en el espacio subaracnoideo.Con la autorregulación abolida la isquemia es inminente, favorecida por el aumento de resistencia debido a la disminución del calibre vascular. La hipertensión endocraneana se explica por la hiperemia en los territorios vasodilatados, espontánea o provocada en forma iatrogénica.El trastorno de la microcirculación puede ser estudiado por el tiempo de circulación cerebral, el test de hiperemia transitoria, la prueba de la acetazolamida y la reactividad al aumento de pCO2. La autorregulación abolida precede a la manifestación del vasoespasmo en varios días.
Este proceso fisiopatológico mecánico es presentado en forma hipotética, con sus implicancias para el tratamiento.
Palabras clave: Hemorragia subaracnoidea y vasoespasmo; HSA y autorregulación; Velocidad de flujo; Déficit isquémico tardío.
SUMMARY
The basal brain arteries are a system of high flow velocity and high shear stress and energy dissipation, with collateral vessels that get profit from those facts, with a low pressure, that could have some influence in the ischemic pathology at this level, and a distal zone where the flow arrives with lower energy, comparatively with extracranial vessels of the same calibre.
It is possible that the increase of flow velocity at the basal arteries after SAH be a consequence of a diminished peripheral resistance due to autoregulation abolition. Because of the principle of energy preservation, the increase in velocity diminishes the lateral pressure producing a passive diameter decrease of the artery, increasing the width of the wall, that increases the vascular tone, and tends to decrease the diameter, closing a noid hemorrhage using a transient hyperemic response test of vicious circle. Other factors are the extraluminal pressure and the presence of clots outside the vessel wall.
With abolished autorregulation, ischemia is impending, facilitated by the increased resistance opposed by the diminished diameter of the vessels. The intracranial hypertension can be explained by the spotaneous or iatrogenic hyperemia.
The microcirculatory trouble proposed can be assessed by the cerebral circulatory time, the transient hyperemia test, the test of acetazolamide and the reactivity to the increase in pCO2. The abolition of autorregulation so displayed, precedes vasospasm for days. This mechanical physiopathological process is presented hypothetically, with its eventual therapeutic consequences.
Key words: Subarachnoid hemorrhage and vasospasm; Autorregulation; Flow velocity; Delayed ischemic deficit.
INTRODUCCIÓN
Hace un par de años, una búsqueda en MEDLINE sobre el vasoespasmo, desde 1953, arrojó la suma de más de 38.000 publicaciones sobre el tema1. Esto podría deberse a varias causas: a) sus características clínicas, sus consecuencias funcionales y su terapéutica todavía son en buena parte desconocidas, b) la inveterada costumbre de publicar resultados similares, confirmando las bondades de un método diagnóstico o terapéutico, c) la proliferación de métodos diagnósticos y terapéuticos frecuentemente caros y poco efectivos, cuando las condiciones fisiopatológicas de un padecimiento son mal conocidas y la estratificación de pacientes pasibles de diferentes tipos de terapia, se basa en principios poco confiables. Es posible que en el caso del vasoespasmo, los tres mecanismos sean la causa de semejante proliferación literaria.
Nuestra intención en estas páginas es revisar algunas creencias que estimamos firmemente instaladas en la población neuroquirúrgica y entre los intensivistas, anestesiólogos, internistas y otros miembros del equipo tratante y analizar con espíritu crítico, y respaldo bibliográfico, la evidencia que las justifica, en un intento de modificar puntos de vista que sostienen actitudes y prácticas que, según nuestra modesta experiencia, pueden ser perjudiciales cuando se aplican en forma protocolizada.
1) Definición
El vasoespasmo como lo concebimos habitualmente es la contracción activa de la pared de un vaso, habitualmente focalizada, ante la presencia de un estímulo adecuado. Esta contracción puede dar síntomas de isquemia en el territorio del vaso afectado, lo que se ha dado en llamar vasoespasmo sintomático. Está claro, que para poder comprobar el vasoespasmo, es necesario realizar un nuevo estudio angiográfico que, por comparación con el previo, muestre1 la estrechez vascular. Como generalmente se reconoce en la angiografía cerebral por cateterismo un método invasivo de cierto riesgo, en la mayoría de los pacientes se prefiere no realizarla, descartando otras causas de empeoramiento como resangrado, hidrocefalia, fiebre, trastornos hidroelectrolíticos y médicos generales, y considerar así que el paciente que presenta un síntoma nuevo más de 48 a 72 hs. después de producida la hemorragia, tiene un "déficit isquémico tardío" o más correctamente un "déficit neurológico tardío" atribuido en la práctica, a un vasoespasmo sintomático1,2.
Se ha reconocido además del vasoespasmo localizado antes descripto, un vasoespasmo difuso, que en determinadas condiciones, sería responsable de síntomas neurológicos generales, como el deterioro de la conciencia, frecuente en estos pacientes.
2) La HSA es la causa del vasoespasmo
Los vasos cerebrales no suelen ser espasmógenos. Hay algunos vasos famosos por su capacidad para espasmodizarse, como la arteria radial, y las coronarias. Por qué los vasos cerebrales presentan esa característica en menor grado, no se conoce bien. Sin embargo, quienes practicamos angiografía recordamos sin duda algún caso en que una inyección defectuosa produjo en la carótida cervical, y eventualmente en algún vaso intracraneano, un vasoespasmo. El contraste inyectado ha sido culpado de algunos casos de vasoespasmo observados3, lo mismo que la presencia de un catéter dentro de la arteria4. Algunos piensan que "el uso decreciente de arteriografía invasiva a favor de alternativas menos invasivas (angioTC, angioRM) pueden alterar la incidencia y severidad del vasoespasmo angiográfico después de la HSA"2. También el trauma quirúrgico normal y otros efectos mecánicos han mostrado ser espasmógenos para los vasos cerebrales2.
No hay duda, sin embargo, fuera de alguna de esas situaciones poco frecuentes, que la asociación HSA-angiografía evidencia vasoespasmo en un período que comienza promediando la primera semana, se acentúa en la segunda para ceder progresivamente a partir de la tercera, y desaparecer después del mes. Es poco común ver vasoespasmo angiográfico en los primeros días posteriores a la HSA. Esto ha llevado a pensar que el que genera la reacción de la pared arterial sería un producto de degradación de la sangre, y actualmente está de moda la oxihemoglobina.
El hecho comprobado de que el vasoespasmo es más probable cuanto mayor es la hemorragia5, con el agregado últimamente de que la presencia de hemorragia ventricular acentuaría el riesgo6,7, ha llevado a la ilusión terapéutica de que la remoción de la sangre de las cisternas durante la operación mejora el pronóstico. Cualquiera que haya comprobado la delicadeza de las membranas cerebrales y el trauma que significa "escarbar" en las cisternas para eliminar coágulos a veces sumamente adheridos, comprenderá que esto no es racional, aunque haya dado resultado en algún caso anecdótico o experimental8. Es notable la reiteración con que se ha repetido este pobre argumento en la literatura internacional. Damos aquí una cita que resume mucho de lo realizado hasta ese momento y que concluye que la disminución de sangre en las cisternas no tiene correlación con una disminución del vasoespasmo9. Tampoco tuvo efecto significativo el uso de rtPA intraoperatorio con el mismo objetivo en un estudio multicéntrico randomizado10 más allá de algunas "tendencias" que exigirían un análisis detallado de los números presentados. El trabajo referido es un ejemplo de que la randomización no siempre arroja resultados parejos en detalles que exigirían una estratificación de los pacientes tratados, como varios de sus comentaristas hacen notar11,12.
Respecto a la influencia de la remoción espontánea de la sangre en la aparición de isquemia cerebral, la bibliografía ofrece opiniones contradictorias13,14, como en cada subtema del vasoespasmo que consideremos.
3) El vasoespasmo aumenta la velocidad de flujo en la arteria
Esto es lógico: si el mismo caudal debe pasar por una sección vascular más estrecha, la velocidad debe aumentar. Este principio simple, está en la base del diagnóstico de vasoespasmo cerebral por ecografía Doppler transcraneana (DPC)15.
Efectivamente, Aaslid y Nornes en 198216 y 198415, establecieron las bases para el uso en la clínica de este medio diagnóstico no invasivo, que durante unos años se utilizó intensamente en los países desarrollados. Una corrección necesaria fue realizada por Lindegaard17, 18 para cambios en la velocidad en la carótida cervical, ya que éstos pueden influir en la de los vasos intracraneanos, estableciendo una relación "normal" intra/extra craneana de hasta 3/1, aunque se asevera que para que el vasoespasmo sea sintomático, el índice debe superar 6. El índice es útil para diferenciar un aumento de velocidad en la cerebral media por aumento del flujo (p. ej. una MA-V), de una disminución del calibre.
Las velocidades de la carótida antes de entrar al cráneo son de 37+ 6,5 cm/seg16. La velocidades de la cerebral media es 62+12 cm/seg, con un rango de 33 a 9016. Aaslid estableció un límite "normal" máximo de 120 cm/seg y valores superiores son considerados patológicos15. Si el índice de Lindegaard, debe superar 6, como pretenden algunos, las velocidades de la cerebral media para el vasoespasmo sintomático, deberían superar largamente los 200 cm por segundo.
El valor del DTC solo, para el diagnóstico del vasoespasmo, ha venido disminuyendo, a medida que la experiencia con su uso se hizo mayor19-21.
"Ante la necesidad de decidir tratar o no tratar, el valor predictivo positivo del DTC debería ser superior al 80% y el valor predictivo negativo de al menos 90%. Sólo velocidades menores de 120 cm/seg o mayores de 200 cm/seg cumplen con estas condiciones, lo que involucra a sólo el 43% de los pacientes con espasmo significativo. Los valores por encima de 120 cm/seg no fueron discriminativos para la ausencia de vasoespasmo significativo, porque los valores predictivos negativos eran similares (alrededor del 75%). Con velocidades menores de 200 cm/seg, el valor predictivo positivo declina marcadamente al 50%, lo que no es clínicamente mejor que tirar una moneda"21.
El uso de DTC tiene algunas limitaciones. Alrededor del 4% de los pacientes presentan una ventana ósea no apropiada para el estudio.
La hipertensión intracraneana, al aumentar la resistencia al flujo, disminuye la velocidad en el Doppler, y aumenta los índices de pulsatilidad y de resistencia (Gossling y Pourcelot respectivamente)22, disminuyendo la velocidad diastólica, y por tanto la media, aunque los picos sistólicos puedan ser algo más altos23.
Si el espasmo está ubicado más allá o más acá del punto donde se mide, los resultados podrían ser confusos24.
Finalmente, los cálculos realizados asumen el flujo laminar, en un tubo rectilíneo, circunstancias que como hemos visto extensamente25, se dan poco en la naturaleza y menos en los vasos cerebrales.
4) Algunos casos paradójicos aceptados en el paradigma
a) La ausencia de vasoespasmo con presencia de síntomas, descartadas otras causas de empeoramiento. Esta circunstancia, y una distribución de los infartos que no siempre se adapta a la anatomía de los grandes vasos, ha hecho sospechar que el vasoespasmo podría darse en vasos de pequeño calibre, no visibles angiográficamente26.
b) La presencia de vasoespasmo no es la sola causa de las lesiones isquémicas: pacientes con vasoespasmo angiográfico severo pueden encontrarse completamente asintomáticos, y drogas que fallan en mejorar el vasoespasmo, como la nimodipina, mejoran sin embargo, el resultado del tratamiento27. La angioplastia, que dilata la arteria espástica, no ha logrado más que resultados anecdóticos en el tratamiento del vasoespasmo28,29, lo mismo que drogas efectivas en relajar el músculo liso del vaso como el verapamilo30. Según este razonamiento, el vasoespasmo sería una condición necesaria pero no suficiente para el desarrollo de la lesión isquémica. La hipotensión durante el curso del proceso agudo, podría aparecer aquí como un cofactor importante.
c) Recientemente, se han reflotado observaciones clásicas de Crompton, respecto a la presencia de infartos en el territorio de vasos perforantes de las arterias de la base, que este autor atribuyó a la compresión de estos vasos por la invasión de la sangre desde el espacio subaracnoideo, al espacio perivascular de Virchow- Robin31. Actualmente, se acepta que la trombosis de pequeños vasos podría actuar como una concausa nada despreciable en la producción de estas isquemias31,32.
El diámetro de los vasos cerebrales
Como estamos tratando de una disminución activa del diámetro de los vasos cerebrales, sería conveniente revisar qué dice la literatura sobre estas dimensiones.
En la tabla 1 mostramos los datos ofrecidos por diferentes autores, que han medido los vasos con diferentes métodos.
Tabla 1. Diámetros de las arterias de la base según diversos autores. Como "Rothon" significamos los trabajos de su grupo (Paullus, Gibo, Gibo, Perlemutter)

Wollschläger et al. inyectaron los cerebros con una modificación de la suspensión de Schlessinger (bariogelatina)33. El grupo de Rothon inyectó "algunas veces" los vasos con acrílico o látex34-36. Gomes inyectó los vasos con resina poliester37 y Gabrielsen y Greitz los midieron sobre las placas de angiografía38 (Fig. 1). Hillen los midió en material cadavérico formolizado, no inyectado, aplastando el vaso entre dos vidrios y midiendo ese ancho con un calibre38 (Fig. 2). Como esa medida representa la mitad de la circunferencia, hemos dividido los datos de este autor por π/2 = 1,57, para obtener el dato del diámetro, que nos permite comparar mejor sus medidas con las de los otros autores.

Fig. 1. Procedimiento de Gabrielsen y Greitz37 para medir los vasos cerebrales en la angiografía reproducido por Weir39. La fórmula que figura debajo, fue aplicada por este último autor para ofrecer una relación corregida de la medida usando los vasos.

Fig. 2. Método utilizado por Hillen38 para medir las arterias transversalmente. La medida representa la mitad de la circunferencia externa del vaso. Dividiendo por π/2, se obtiene el valor del diámetro externo.
Podemos observar que las medidas en vivo y las de los vasos inyectados son entre un 11 y un 24% mayores, que las de los vasos simplemente formolizados. En las medidas radiológicas hay un factor de agrandamiento, ya que las distancias foco- placa eran de 75 u 80 cm, por lo que deberían ser divididas por un coeficiente aproximado a 1,3.
DISCUSIÓN
El paradigma del vasoespasmo está sostenido en varias evidencias y una causa.
1) La visualización angiográfica.
2) El aumento de velocidad en la arteria medido con el DTC.
3) Las lesiones isquémicas en los territorios vasoespásticos.
4) Una causa: la oxihemoglobina o algún otro factor, aportado por la sangre, al espacio subaracnoideo.
- La velocidad de la sangre en las arterias. ¿Qué estamos midiendo?
Hemos visto los valores que hallaron Aaslid et al para la velocidad en la cerebral media, anterior, y carótida interna que han sido confirmados por otros autores. El grupo de Fahlbusch encuentra incluso que, a unos 3 mm del origen de la cerebral media, la velocidad alcanza a 70cm/seg40.
El mismo Aaslid se sorprende de estos valores15. En más de 50% de los casos, la velocidad pico sistólica en la cerebral media llega a 1m/seg, la misma que la velocidad sistólica en la aorta.
La velocidad es mayor que en la carótida interna cervical, que no pasaría de 44 cm/seg, según sus propios datos.
Es un principio fisiológico bien conocido, basado en la ley de la conservación de la energía, que la velocidad de la sangre en cada vaso, desciende progresivamente a medida que nos alejamos del corazón, ya que aunque el diámetro del vaso que estamos observando es menor que el de la aorta o la carótida, por ejemplo, la superficie de la sección vascular total, es decir la suma de las secciones de todos los vasos a ese nivel es mucho mayor que la de los grandes vasos. Aunque en las arterias estas diferencias no son muy grandes, la velocidad en un vaso de menor calibre no puede ser mayor que en uno más grande. Hemos visto sin embargo que para la cerebral media se aceptan valores hasta tres veces mayores que los de la carótida cervical, según el índice de Lindegaard, que causaron la sorpresa de Aaslid y le hicieron aseverar que "en el sujeto normal, las mayores velocidades de la circulación cerebral se encuentran en las arterias de la base"15. En las ramas de la cerebral media, las velocidades son menores que en el tronco, lo que es fisiológicamente correcto.
¿Cuál podría ser el origen de esta paradoja a nivel de la base? ¿Por qué no nos hemos hecho antes esta pregunta?
- La disminución de calibre de la carótida.
En su travesía por la base del cráneo, la carótida no emite ramas importantes, por lo que podríamos considerar despreciable el caudal que le restan al del vaso madre. Podemos considerar por consiguiente, que el caudal de la carótida es el mismo a la entrada de la base que a su salida, a nivel del proceso clinoideo.
El calibre del vaso, sin embargo ha disminuido notoriamente. Si tomamos los valores de la tabla I, podemos asignar a la entrada a la base, un radio de 5,6 mm y antes de la emergencia de la oftálmica, de 4 mm. El valor de las respectivas superficies de sección, es de 98,47 mm2 y 50,24 mm2. Por lo tanto, la velocidad de la corriente sanguínea aumentaría prácticamente al doble, unos 80 a 88 cm/seg. Este hecho tendría un efecto muy importante. Primero, al aumentar la energía cinética, disminuye la presión lateral por el principio de Bernouilli. Segundo, aumenta el rozamiento (shear stress) de las líneas de corriente entre sí, y de todo el conjunto con las paredes de los vasos, lo cual tiende a disminuir la energía de la masa circulante.
A partir de este momento, la carótida da varias ramas importantes que nacen de su tronco en forma perpendicular, lo que tiende a disminuir la energía cinética en los vasos colaterales, aprovechando la disminución de la presión lateral en el vaso madre.
Posiblemente, la velocidad en la carótida disminuya rápidamente al dar estas ramas. Los datos de la bibliografía no abundan, porque por su disposición espacial, el ángulo de incidencia del haz de ultrasonido es casi perpendicular, haciendo más inexactos los cálculos15,16.
- Factores circulatorios.
La cerebral media es la continuación anatómica de la carótida intracraneana, y posiblemente reciba todo su momento (energía cinética), sin embargo de acuerdo a la ley de la conservación de la energía, la velocidad en ella debería disminuir, como explicamos antes. Aparentemente, no sucede así, y la velocidad media en la raíz de la silviana alcanza valores similares a los de la carótida intracraneana.
Si tomamos el estudio del grupo Fahlbusch, a 3 mm del origen, la velocidad en la cerebral media alcanza a 70 cm/seg como promedio, llegando hasta los 150 cm/seg40.
Es posible que en esta región se estén formando los remolinos de von Karman que hemos estudiado antes25 y que se trate de un artificio generado por ellos, más que de una velocidad verdadera, que podría ser entonces, algo menor.
En resumen, en la base del cerebro tenemos un sistema de alta velocidad y de gran disipación de energía de la masa circulante por rozamiento (flujo alterado), con un sistema de vasos colaterales que se benefician de estos fenómenos, con una baja presión, que podría tener importancia en la patología isquémica a este nivel; y una zona distal, a la que la corriente sanguínea llega con menor velocidad y presión, comparativamente con los vasos extracraneanos del mismo calibre25,41.
- La regulación de la velocidad en los vasos de la base del cerebro.
Contrariamente a lo que pensamos, la velocidad de flujo en las arterias de la base no se debe solamente a la variación del diámetro de los mismos. Una variación de la resistencia periférica tiene el mismo efecto42. Variaciones de la resistencia periférica deben ser muy comunes en el cerebro, porque ya estamos acostumbrados a ver las imágenes funcionales que nos ofrecen el PET y la IRMf, en donde las áreas cerebrales puestas en juego para una tarea, aumentan su circulación, probablemente por aumento de los requerimientos metabólicos. Así que es posible que las velocidades en estos vasos se modifiquen en forma continua, tanto durante la vigilia como durante el sueño, según las cambiantes actividades del cerebro.
La hipertensión endocraneana, que aumenta la resistencia al flujo, disminuye la velocidad.
- En busca de una concepción "pasiva" del vasoespasmo.
Muchos autores coinciden en que la HSA produce una abolición de la autorregulación45-48, ya sea territorial o generalizada. Conocemos el significado del término: incapacidad para preservar la constancia del flujo ante diferentes presiones de perfusión. O su inversa, dependencia absoluta del flujo, de los valores de presión de perfusión.
¿Cómo nos imaginamos que están los vasos de resistencia (arteriolas) en un territorio con su autorregulación abolida? No hay otra manera que pensar que están vasodilatados, porque de estar contraídos, ofrecerían una resistencia al flujo con los cambios de presión.
Siguiendo con nuestro razonamiento del punto anterior, una disminución de la resistencia periférica de esta causa, produciría un aumento de la velocidad circulatoria secundario en los grandes vasos de la base, o en alguno de ellos, de ser regional.
El aumento de la velocidad circulatoria en estos vasos, producirá, por la ley de conservación de la energía, una disminución de la presión lateral en una proporción similar a la del aumento de la energía cinética. Debido a ello, el vaso disminuirá su calibre. La disminución de calibre, tendrá dos efectos: uno, sobre la pared, que aumentará su espesor. Este aumento de espesor, producirá un aumento del tono vascular (ver parte II de nuestra saga49), que tenderá a disminuir aún más el diámetro del vaso. Por otro lado, al aumentar la velocidad por disminución del calibre, la presión lateral disminuirá aún más, realimentando esta especie de círculo vicioso. Visto angiográficamente, este cuadro podría ser interpretado fácilmente como vasoespasmo. Es posible que este mecanismo alcance para producir una disminución de al menos un 25% del calibre del vaso, teniendo en cuenta los datos con que contamos de la tabla 1. Una arteria sin circulación después de la muerte, se colapsa aproximadamente en esa proporción. El resto debería hacerlo el aumento de tono vascular, que en la arteria de cadáver carece de la acción de uno de sus integrantes a saber, el músculo liso.
El efecto de la presión externa.
El vaso está inmerso en la cavidad intracraneana, que tiene una presión positiva de valor variable, pero posiblemente mayor de cero, después de una HSA, por lo que tenderá a actuar de forma concéntrica, deformando la pared del vaso en el sentido de una disminución mayor del calibre. Es más, la presencia de coágulos a cierta tensión en el espacio subaracnoideo, que ha sido propuesta como causa del estrechamiento vascular en la HSA, podría también jugar algún rol, especialmente en las estrecheces más localizadas.
1) Consecuencias de la hipótesis
Tendremos que sacar algunas consecuencias de esta hipótesis y enfrentarlas a los hechos, para verificarla o rechazarla.
1) Según esta hipótesis, los vasos de conducción deberían presentar una disminución pareja del calibre en todo su recorrido. Es decir, no debería haber espasmo localizado.
Newell et al. publicaron en 1990 un estudio sobre la distribución del vasoespasmo angiográfico50, y observaron que en el 50% de los pacientes con un vasoespasmo igual o mayor que 25%, éste estaba localizado en los troncos proximales; en un 42,5%, el espasmo involucraba tanto las porciones proximales, como las distales del vaso, y en el 7,5%, el espasmo era distal.
Si estos datos son fidedignos, al menos en cerca de la mitad de los casos, se podría invocar el mecanismo que preconizamos. Otros autores han hecho observaciones parecidas con proporciones diversas.
En este sentido, es interesante la observación de Matzusaki et al. que observaron que el defecto isquémico tardío ocurría en sus pacientes cuando el "estrechamiento suave" de los vasos afectaba simultáneamente a M1 y M251. El original de este trabajo está en japonés, por lo que lamentablemente podemos reproducir sólo datos del resumen.
2) Consecuencias de la vasodilatación de los vasos de resistencia.
Según esta hipótesis, en los pacientes con vasodilatación periférica cerebral por abolición de la autorregulación, debería haber un enlentecimiento de la circulación cerebral al entrar la sangre en una red ampliamente abierta de pequeños vasos francamente dilatados y, a la vez, un aumento del volumen sanguíneo cerebral. Esto llevará a un aumento de la extracción de oxígeno, y a una tasa de metabolismo del oxígeno dependiente de las fluctuaciones del flujo sanguíneo cerebral con los cambios de presión de perfusión.
En 1977 Grubb et al publicaron un estudio en el que encuentran un aumento significativo del volumen sanguíneo cerebral y de la tasa de extracción de oxígeno, junto con una disminución del flujo sanguíneo cerebral y por ende de la tasa metabólica de oxígeno. Su notable conclusión es que "el vasoespasmo cerebral consiste en constricción de los vasos extraparenquimatosos grandes, radiológicamente visibles, acompañada de una dilatación masiva de los vasos intraparenquimatosos"52. También hay observaciones de un caso53 en donde la vasodilatación (vasoparálisis) precedió en varios días al vasoespasmo sintomático.
En 2003, Minhas et al.54 estudiando con PET 25 pacientes con déficit neurológico tardío encontraron variaciones importantes en el flujo cerebral, desde hipoflujo hasta hiperemia pasando por valores normales. Los valores de Doppler transcraneano y el índice de Lindegaard no se correlacionaron significativamente ni con los déficits ni con el flujo cerebral. El flujo fue algo menor en pacientes fallecidos que en los que sobrevivieron, pero en los fallecidos, el promedio estuvo bastante por encima de los valores de isquemia (32,3 ml/100g/min y 36,0 ml/100g/min respectivamente. Debemos tener en cuenta que todos estos pacientes estaban siendo tratados con triple H. Los autores concluyen que "el concepto clásico de vasoespasmo causando isquemia cerebral tardía está sobresimplificado". De un parecer similar, parecen haber sido Clyde et al.55
El tiempo de circulación cerebral se puede estudiar mediante la inyección de radioisótopos56,57 o angiográficamente56.
Éste es igual a la razón entre el volumen sanguíneo cerebral global y el flujo cerebral global57. Diferentes autores58,59 han aplicado el método a los pacientes con HSA aneurismática. Se ha encontrado que una prolongación del tiempo de circulación cerebral es pronóstico de vasoespasmo cuando el estudio es realizado en los primeros días, en los que el diámetro de los vasos de la base es todavía normal58,59. Este hecho nos hace reflexionar acerca de cuál sería el desarrollo de los acontecimientos desde el punto de vista de nuestra hipótesis.
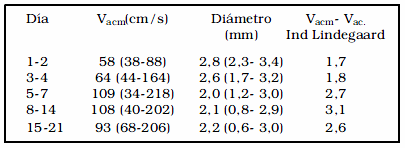
En la tabla 2, tomada de Lindegaard17, vemos que se trata de un grupo heterogéneo de pacientes, muchos de los cuales no deben de haber desarrollado vasoespasmo, y otros sí, posiblemente los menos, teniendo en cuenta los valores de la velocidad promedio. Se puede observar que quienes tendrán "vasoespasmo" (velocidades máximas en cada grupo), están comenzando el primer día con el desarrollo del círculo vicioso descripto (disminución de la resistencia periférica). Estos pacientes tendrán posiblemente desde estos tempranos momentos, una prolongación del tiempo circulatorio, con diámetros angiográficos aproximadamente normales.
Tabla 2. Curso temporal de velocidades y diámetros de la arteria cerebral media en 51 pacientes con HSA, tomado de Lindegaard17.
3) La explicación de la isquemia.
La falta de autorregulación expone a los territorios afectados a una situación de isquemia inminente. La misma se desencadenará siempre que la presión de perfusión alcance unos límites inferiores, que dependerán del paciente y las circunstancias. Hipovolemia, hiponatremia, hipotensión, disminuirán el flujo cerebral eventualmente hasta esos límites.
La disminución de calibre de los vasos de la base, por su lado, aumentará fuertemente la resistencia al flujo, favoreciendo el fallo circulatorio.
Apenas la disminución del flujo alcance niveles de penumbra, el paciente se pondrá sintomático, pero no necesariamente en forma irreversible, y si bien las lesiones observadas en la tomografía tendrán una distribución correspondiente a la del territorio o territorios afectados, podrán desaparecer en algún caso, o disminuir considerablemente su extensión en otros, si las medidas de sostén del flujo llegan a tiempo.
El hecho que el "vasoespasmo sintomático" (déficit isquémico tardío) sea de aparición tardía, se debe posiblemente, entre otros factores, a que los procesos que llevan a la hipovolemia requieren de varios días para establecerse y ser efectivos60.
En los vasos perforantes se debe dar un fenómeno especial por el cual éstos están más sujetos a isquemia, no solo en el caso de la hemorragia subaracnoidea1. Estos vasos nacen en forma perpendicular al vaso madre, y cuanto más gruesos, más pronto adoptan un trayecto recurrente, es decir, la corriente sanguínea en ellos lleva una dirección opuesta a la del vaso madre. Con el aumento de velocidad en éste, es posible que estos vasos sean los primeros afectados hemodinámicamente por la hipotensión debido a su especial disposición (Fig. 3). Como algunos de ellos irrigan territorios importantes en el mantenimiento del estado de vigilia, no será pues extraño que una alteración del mismo sea uno de los síntomas cardinales del "vasoespasmo sintomático".

Fig. 3 Trayecto de los vasos perforantes de la arteria cerebral media normal en una paciente de 25 años. Obsérvese el trayecto recurrente de las lentículo estriadas laterales que son las más gruesas.
4) Explicación de la hipertensión endocraneana.
Dejando de lado los fenómenos de bloqueo de la circulación del LCR por la hemorragia, se pueden invocar dos mecanismos para esta situación nada infrecuente después de HSA:
a) el aumento del volumen sanguíneo cerebral.
Cuando los territorios afectados por la falta de autorregulación son extensos, el aumento del volumen sanguíneo cerebral puede producir una "hinchazón" cerebral difusa, causa del aumento de la PIC siempre y cuando lleve a los fenómenos de desplazamiento y hernia61.
Este caso se daría en algunos pacientes que entran con peor grado clínico, con una HSA grado III o IV.
b) Hiperemia secundaria a aumento de la presión de perfusión.
Un aumento de la presión arterial afectará a los territorios vasodilatados, produciendo un aumento del flujo que, de acuerdo a su magnitud, provocará un aumento aún mayor del volumen sanguíneo cerebral en ellos, o un cuadro de edema progresivo, desencadenando desplazamiento y hernia, y la consiguiente hipertensión endocraneana.
Como dicen Clyde et al, velocidades aumentadas en el Doppler, pueden indicar también perfusión aumentada y no sólo un compromiso en la perfusión causado por estrechamiento arterial54.
Muchos pacientes con "vasoespasmo sintomático" tratados con triple H terminan su vida, quizá, de este modo, al pasar de una situación de oligohemia a una de hiperemia iatrogénica.
La triple H ha dejado de ser recomendada recientemente62 en forma preventiva, aunque es utilizada como protocolo por casi todos los Servicios de Terapia Intensiva y es considerada evidencia "razonable" para el tratamiento del vasoespasmo (Clase II, nivel de evidencia B) en las "Guidelines" de marzo de 2009.
Debemos hacer notar que la hipertensión endocraneana aumenta la resistencia al flujo y por ende, disminuye la velocidad circulatoria en los vasos de la base.
- Puntos oscuros en esta hipótesis.
a) El espasmo angiográfico localizado. Este es el punto de arranque de nuestras creencias actuales acerca del vasoespasmo, y se trata de una evidencia en todo el sentido de la palabra: es visible, y repetiblemente visible.
Hemos visto sin embargo, que es la asociación de angiografía con HSA la que muestra frecuentemente y luego de un período de algunos días, la alteración del vaso. Hemos visto que hay autores que suponen que es posible que cuando se reemplace la angiografía directa por métodos no invasivos, este tipo de espasmo se reduzca notablemente.
¿Hay derecho a pensar así? Si bien la inyección angiográfica genera una serie de alteraciones de la presión sistémica63 y local64 y de la velocidad circulatoria homo y contralateral65, que podrían provocar variaciones del calibre en un sistema con el tono vascular aumentado, ya nos encontramos en una etapa del desarrollo de la TAC, que permite visualizar el angioespasmo. Exámenes multislice realizados antes de la angiografía digital, presentan una correlación muy buena con ésta tanto para el diagnóstico del vasoespasmo como para la negación de su existencia. En esta etapa inicial, sin embargo, no hay todavía interés en conocer el tipo de espasmo que presentan los vasos, y el análisis se ha hecho en términos de la cantidad de "segmentos vasculares" espásticos o no, sobre 251 examinados, en un total de 17 pacientes. Como se encontraron 74 segmentos espásticos y 40 "hemodinámicamente significativos", tenderíamos a pensar que predominó el espasmo difuso en esta serie66.
Las estenosis localizadas pueden ser en algunos casos placas de ateroma4,31, compresión extrínseca por coágulos subaracnoideos, y en otros, verdadera estenosis vascular "espástica".
- Consecuencias para el tratamiento.
La hipótesis explica muy bien el efecto deletéreo de la hipotensión sobre estos pacientes. La hipotensión es un evento bastante posible en el paciente con HSA, por hiponatremia, disminución de la volemia por aporte insuficiente, por efecto de drogas anestésicas o depresores del sistema nervioso central y por la misma nimodipina, como veremos. Evitar la hipotensión y la hipovolemia debe ser una preocupación central en el tratamiento de estos enfermos60.
Si nuestra hipótesis es correcta, la hiperdinamia podría ser perjudicial en algunos casos, sobre todo aquellos en que la autorregulación se encuentra alterada en áreas extensas, o cuando ya se ha producido infarto67, por lo que protocolos que la incluyan como primer medida deberían ser tomados con pinzas. Reponer la volemia normal y la TA normal, puede ser suficiente en muchos casos, sobre todo en aquellos que desarrollan un cuadro focal, o multifocal, con focos pequeños de isquemia en la TAC. Si la situación no revierte, el uso de medidas de hiperdinamia será efectivo en algunos y no en otros. En estos últimos, es posible que se haya producido un infarto, y la hiperdinamia será no sólo inefectiva, sino eventualmente peligrosa. Finalmente, en pacientes con lesiones más extensas, que involucran territorios centrales, la hiperdinamia será en extremo riesgosa, con más probabilidad de hinchazón y edema cerebral de reperfusión68 con alto riesgo de hipertensión endocraneana. En general, en estos casos, la operación descompresiva no ofrece buenos resultados según nuestra experiencia.
Resulta finalmente irracional continuar tozudamente con las medidas 3H cuando el paciente presenta hipertensión endocraneana debido a la presencia de un gran foco de edema pos infarto cerebral.
Tan tempranamente como 1993, Shimoda67 llamó la atención acerca de los peligros de la 3H en pacientes con lesiones extensas y grado alto en la escala clínica de HSA. Además, con gran cordura, habló de 3H estándar y 3H "óptima". No había diferencias entre las medidas tomadas en una u otra, pero en la óptima, éstas eran suspendidas en el mismo momento en que los síntomas revertían o aparecían signos de infarto en la TC. Esta práctica fue la que obtuvo los mejores resultados.
El objetivo terapéutico debería ser el de alcanzar una perfusión mínima adecuada, y no forzar la dilatación de los vasos de la base. Si este objetivo se cumple, es posible que las velocidades en el DTC sean muy altas. No deberían traer preocupación, si el cuadro clínico revirtió.
No sólo Shimoda, sino muchos autores llamaron la atención acerca de los riesgos de la 3H: edema cerebral, hipertensión endocraneana, insuficiencia cardíaca, edema agudo de pulmón, sepsis, taquiarritmia, etc69
- El huevo o la gallina.
Todos los autores coinciden y remarcan que la HSA puede generar vasoespasmo. Muchos coinciden también en que produce alteración de la autorregulación. Otros piensan que la vasodilatación periférica es una respuesta autorreguladora al vasoespasmo proximal52. Pocos han asociado el estudio o la evaluación del vasoespasmo con la alteración de la autorregulación. Un trabajo interesante del grupo de Pikcard y Czosnika, en Cambridge, Inglaterra, muestra una correlación estrecha entre intensidad del vasoespasmo e intensidad en el desarreglo de la autorregulación70. Este mismo grupo, demostró que el desarreglo autorregulatorio precede en varios días al desarrollo de vasoespasmo y déficits isquémicos tardíos en el 100% de los casos, por lo que su testeo mediante la prueba de hiperhemia transitoria tendría valor pronóstico71,72.
Lo que cabe preguntarse entonces, es si la alteración de la autorregulación no es la primera causa del desarrollo de los eventos circulatorios posteriores a la HSA.
- La naturaleza del trastorno autorregulador.
Lo más notable de este trastorno, es que no se trata de una vasoparálisis, ya que los vasos presentan una buena respuesta constrictora al anhidrido carbónico70. La hiperventilación produce una disminución significativa de la velocidad de flujo en los vasos de la base70.
He aquí una posibilidad terapéutica que no ha sido profundamente analizada hasta el momento: hiperventilación moderada, manteniendo una buena presión de perfusión. La vasoconstricción de la hipocarbia equilibra la eventual hiperhemia por hiperdinamia.
En cambio, el aumento en 6 mmHg. de la pCO2 por encima de los valores basales, no produce ningún efecto73. Esto podría deberse, según nuestra hipótesis, a que los vasos están ya dilatados y no pueden dilatarse más por la acción del CO2. En esto, el trastorno microcirculatorio, se parecería a la vaso parálisis.
En un estudio reciente, todos los pacientes que desarrollaron vasoespasmo sintomático, presentaron previamente en un promedio de 4 días, una falta de reactividad al estímulo con CO273 (sensibilidad 100%). Ningún paciente con reactividad normal, presentó vasoespasmo sintomático (valor predictivo negativo 100%). Algunos pacientes sin vasoespasmo sintomático presentaron reactividad anormal (especificidad 55%). Por las velocidades registradas en estos últimos pacientes en el DTC (> 120 cm/seg), es posible que hayan tenido "vasoespasmo" asintomático.
La falta de respuesta a la acetazolamida53 puede constituir otra muestra precoz del trastorno autorregulador.
- El aumento del tono vascular y la participación del músculo liso.
a) Hay tanta bibliografía publicada acerca del tema, que parece muy arriesgado contradecirla, en cierta manera. Nuestra hipótesis sostiene efectivamente, que existe en el vasoespasmo, un aumento del tono vascular, pero éste es secundario a un trastorno hemodinámico generado en la microcirculación.
Lo que la hipótesis no niega, es su participación en el aumento de la resistencia vascular, favoreciendo la amenaza de isquemia producida por el trastorno primario, que es la alteración de la autorregulación.
Como hemos estudiado49, el tono vascular depende de la geometría de la pared (a más espesor más tono) y de la estructura de la misma: grosor de la capa de músculo liso y proporción elastina-colágeno.
En la arteria cerebral tenemos una capa de músculo liso, y un espesor exiguos49. Es posible que el músculo de los vasos de la base responda a mecanismos neurales, en vista de su profusa inervación. Todos estos factores nos hacen pensar que la participación del músculo liso en el vasoespasmo de la HSA, si bien existe, no sea el mecanismo determinante de isquemia.
b) Los vasodilatadores. Desde que el más antiguo
de los autores de este trabajo se inició en la práctica neuroquirúrgica, han pasado muchas sustancias "farmacológicamente activas" contra la contracción del músculo liso arterial. Hydergina, kanamicina, hidroxifilina, papaverina, nicardipina, nimodipina, verapamilo, sulfato de magnesio, entre las más usadas. Por ahora, permanece la nimodipina, con un efecto modesto2, pero aparentemente indudable sobre el resultado.
- El efecto de la nimodipina.
Es reconocido el efecto positivo de la nimodipina en el resultado de la HSA. Sin embargo, es también corriente el concepto que esta mejoría no se debe a su acción relajante del músculo arterial, ya que no hay prueba fehaciente de que actúe sobre el vasoespasmo. Así, se ha llegado a la conclusión de que actúa por un efecto directo protector de la neurona27.
Pero la nimodipina es un vasodilatador. ¿Actuará sobre la musculatura de los pequeños vasos? Si es así, ¿qué efecto tendrá sobre un vaso dilatado por pérdida de su capacidad autorreguladora?
Lo más probable es que no tenga ningún efecto, pero podría darse que, como el CO2, tenga un efecto independiente, produciendo una vasodilatación, que acentúe la posibilidad de aparición, o la magnitud del vasoespasmo. O que al dilatar territorios autorregulados, produzca un "robo" en el área no autorregulada.
Aunque se la presentó como un vasodilatador cerebral selectivo, tiene efectos generales, aún por vía oral, generando a veces hipotensión, que podría resultar perjudicial y que debe ser revertida rápidamente.
- La papaverina intraarterial.
Se ha demostrado desde la década del 90 en numerosos trabajos que la papaverina liberada en los vasos cerebrales mediante cateterismo selectivo, tiene efecto benéfico sobre el vasoespasmo, produciendo dilatación vascular comprobable, junto con mejoría de los síntomas en muchos casos (aproximadamente 50%). Sin embargo, su efecto suele ser transitorio, regresando en 12 a 24 hs, por lo que se requieren intervenciones sucesivas.
Por otro lado, no está exenta de riesgo: hipertensión endocraneana, necrosis cortical, convulsiones, ceguera, midriasis, entre las más nombradas74-76. La papaverina parecería no tener efecto benéfico sobre el resultado final.
- La angioplastia
Esta arma terapéutica produce una dilatación vascular forzada. No se trata de vencer la resistencia del músculo "espástico", sino de alterar a veces marcadamente la estructura de la pared arterial76, que se mantendrá dilatada por un largo período. Desde este punto de vista, es una terapéutica antifisiológica que, al aumentar el diámetro vascular, puede disminuir la resistencia al flujo como para mejorar algunos casos selectos de isquemia por "vasoespasmo".
Tendría entonces, según nuestra opinión, una indicación limitada a vasos grandes de la base con vasoespasmo localizado, en casos de isquemia incipiente. Simultáneamente tiene riesgos graves, como ruptura, disección arterial, tromboembolismo, hemorragia de reperfusión y necesidad de anticoagulación76.
CONCLUSIÓN
Hemos presentado una hipótesis mecánica, basada en datos bibliográficos, que parte del principio que el vasoespasmo es la consecuencia de un trastorno en la microcirculación cerebral causado por la HSA. La primacía temporal del trastorno autorregulador sobre la aparición de la constricción vascular, ha sido demostrada por diferentes autores mediante diferentes métodos, como hemos visto. La incapacidad para producir una respuesta hiperhémica adecuada a diversos estímulos y la prolongación del tiempo circulatorio cerebral preceden en días u horas, al desencadenamiento de vasoespasmo asintomático o de los síntomas de daño neurológico tardío.
A partir de este principio, hemos podido explicar complejos efectos sobre la circulación cerebral que han sido observados por distintos autores, y especialmente, la isquemia y la hipertensión endocraneana que son causa frecuente de muerte o daño funcional irreversible en esta patología.
Las consecuencias para el tratamiento han sido discutidas proponiendo alguna alternativa, y que las medidas de protocolo sean adaptadas a cada paciente en cada circunstancia.
El fracaso rotundo y sistemático de los vasodilatadores más potentes para relajar la contracción arterial, y el riesgo en que ponen al paciente cuando lo logran, es un argumento en contra de que el vasoespasmo sea un proceso causal primario, en donde el músculo liso de las arterias de la base sea el protagonista principal.
Como esta presentación es hipotética, muchos de estos hechos descriptos requerirán un análisis a la luz de un conocimiento mayor, o de la producción de evidencia experimental o clínica superior a la actual, que tenga por objetivo aceptarla o rechazarla.
1. Stein SC, Levine JM, Nagpal S, LeRoux PD. Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurg Focus, 2006; 21(3): E2. [ Links ]
2. Nolan, CP, Macdonald RL. Can angiographic vasospasm be used as a surrogate marker in evaluating therapeutic interventions for cerebral vasospasm? Neurosurg Focus, 2006; 21(3): E1. [ Links ]
3. Yasargil MG. Microneurosurgery. Vol. I. pp 196. Georg Thieme Verlag, Stuttgart. Thieme Stratton Inc. New York, 1984. [ Links ]
4. Morris P. Practical neuroangiography. Wilkins & Williams, Baltimore, 1997. [ Links ]
5. Fischer CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomografic scanning. Neurosurgery, 1980; 6: 1-9. [ Links ]
6. Frontera JA, Claassen J, Michael Schmidt J, Wartenberg KE, Temes R, Sander Connolly E, et al. Prediction of symptomatic vasospasm after subarachnoi hemorrhage : the modified Fischer scale. Neurosurgery, 2006; 59: 21-7. [ Links ]
7. Klimo P, Schmidt RH. Computed tomography grading schemes used to predict cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a historical review. Neurosurg Focus, 2006; 21(3): E5. [ Links ]
8. Mayberg MR. Intracranial arterial spasm. En: Wilkins RH, Rengachary SS (Eds.) Neurosurgery, Mc Graw-Hill, New York, Tomo II, 1966. [ Links ]
9. Inagawa T, Yamamoto M, Kamiya K. Effect of clot removal on cerebral vasospasm. J Neurosurg, 1990; 72: 224-30. [ Links ]
10. Findlay JM, Kassell NF, Weir BKA, Haley EC, Kongable G, Germanson T, et al. A randomized trial of intraoperative, intracisternal tissue plasminogen activator for the prevention of vasospasm. Neurosurgery, 1995; 37: 168-78. [ Links ]
11. Loftus CM. "Comment" de cita 10.
12. Batjer HH. "Comment" de cita 10.
13. Browers PJAM, Wijdicks efm, Van Gijn J. Infarction after aneurysm rupture does not depend on distribution or clearance rate of blood. Stroke, 1992; 23: 374-9. [ Links ]
14. Reilly Ch, Amidei Ch, Tolentino, J, Jahromi BS, Macdonald, RL. Clot volume and clearance rate as independent predictors of vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg, 2004; 101: 255- 261. [ Links ]
15. Aaslid R, Huber P, Nornes H. Evaluation of cerebrovascular spasm with transcranial Doppler ultrasound. J Neurosurg, 1984; 60: 37- 41. [ Links ]
16. Aaslid R, Markwalder T-M, Nornes H. Noninvasive transcranial Doppler ultrasound recording of flow velocity in basal cerebral arteries. J Neurosurg, 1982; 57: 769-74. [ Links ]
17. Lindegaard K-F, Nornes H, Bakke SJ, Nakstad P. Cerebral vasospasm after subarachnoid haemorrhage investigated by means of transcranial Doppler ultrasound. Acta Neurochir, 1988 (Supp 42); 81-4. [ Links ]
18. Lindegaard K-F, Nornes H, Bakke SJ, Sorteberg W, Nakstad P. Cerebral vasospasm diagnosis by means of angiography and blood velocity measurements. Acta Neurochir, 1989; 100: 12-24. [ Links ]
19. Vora, Y Y, Suarez-Almazor, M, Steinke, DE, Martin,ML, Findlay, JM. Role of transcranial Doppler monitoring in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery, 1999; 44: 1237-47. [ Links ]
20. Eckelund, A, Säveland H, Romner B, Brandt L. Is transcranial Doppler sonography useful in detecting late cerebral ischaemia after aneurysmal subarachnoid hemorhage? B J Neurosurg, 1996; 10: 19-25. [ Links ]
21. Creissard P, Proust F, Langlois O. Vasospasm diagnosis : theoretical and real transcranial Doppler sensitivity. Acta Neurochirurgica, 1995; 136: 181-5. [ Links ]
22. Klingelhöffer J, Sander D, Holzgraefe M, Bischoff Ch, Bastian C. Cerebral vasospasm evaluated by transcranial Doppler ultrasonography at different intracranial pressures. J Neurosurg, 1991; 75: 752-8. [ Links ]
23. Okada Y, Shima T, Nishida M, Kanji Y, Takashi H, Chie Y, Akira Y. Comparison of transcranial Doppler investigation of anerysmal vasospasm with digital subtraction angiographic and clinical findings. Neurosurgery, 1999; 45: 443-50. [ Links ]
24. Fontana H, Belziti H, Requejo F, Buratti S, Recchia M. La circulación cerebral en condiciones normales y patológicas. Parte III. El flujo sanguíneo en los vasos de la base. Rev Arg Neuroc, 2007; 21: 187-99. [ Links ]
25. Okuma H, Manabe H, Tanaka M, Suzuki S. Impact of cerebral microcirculatory changes on cerebral blood flow during cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke, 2000; 31: 1621-7. [ Links ]
26. Mesis RG, Wang H, Lombard FW, Yates R, Vitek, MP, Borel CO, et al. Dissociation between vasospasm and functional improvement in a murine model of subarachnoid hemorrhage. Neurosurg Focus, 2006; 21(3): E4. [ Links ]
27. Muizelaar JP, Zwienenberg M, Rudisill NA, Hecht ST. The prophylactic use of transluminal balloon angioplasty in patients with Fischer grade 3 subarachnoid hemorrhage: a pilot study. J Neurosurg, 1999; 91: 51-8. [ Links ]
28. Mazumdar A, Rivet DJ, Derdeyn CP, Cross DWT, Moran C. Effect of intraarterial verapamil on the diameter of vasospastic intracranial arteries in patients with cerebral vasospasm. Neurosurg Focus, 2006; 21(3): E15. [ Links ]
29. Polin RS, Coenen VA, Apperson Hansen C, Shin P, Baskaya MK, Nanda A, Kassell NE. Efficacy of transluminal angioplasty for the management of symptomatic cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg, 2000; 92: 284- 90. [ Links ]
30. Crompton MR. The pathogenesis of cerebral infarction following the rupture of cerebral berry aneurysms. Brain, 1964; 87: 491-510. [ Links ]
31. Rabinstein AA, Weigand S, Atkinson JLD, Wijdicks EFM. Patterns of cerebral infarction in aneurysmal subachnoid hemorrhage. Stroke, 2005; 36: 992-7. [ Links ]
32. Stein SC, Levine JM, Nagpal S, LeRoux PD. Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurg Focus, 2006; 21(3): E2. [ Links ]
33. Wollschlaeger g, Wollschlaeger PB, Lucas FV, Lopez VF. Experience and result with postmortem cerebral angiography performed as routine procedure of the autopsy. Am J Roentgenol, 1967; 101: 68-87. [ Links ]
34. Gibo H, Lenkey C, Rothon ALJr. Microsurgical anatomy of the supraclinoid portion of the internal carotid artery. J Neurosurg, 1981; 55: 560-74. [ Links ]
35. Gibo H, Carver Ch, Rothon ALJr, Lenkey, C, Mitchell RJ. Microsurgical anatomy of the middle cerebral artery. J Neurosurg, 1981; 54: 151-69. [ Links ]
36. Paullus WS, Pait G, Rothon ALJr. Microsurgical exposure of the petrous portion of the carotid artery. J Neurosurg, 1977; 47: 713- 26. [ Links ]
37. Gomes FB, Dujovny M, Umansky F, Berman SK, Diaz FG, Ausman JI, et al. Microanatomy of the anterior cerebral artery. Surg Neurol, 1986; 26: 129-41. [ Links ]
38. Gabrielsen TO, Greitz T. Normal size of the internal carotid, middle cerebral and anterior cerebral arteries. Acta Rad (Diagn), 1970; 10: 1-10. [ Links ]
39. Hillen B. The variability of the circle of willis: univariate and bivariate analysis. Acta Morphol Neerl-Scand, 1986; 24: 87-101. [ Links ]
40. Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J Neurosurg, 1978; 48: 173-8. [ Links ]
41. Laumer R, Steinmeier R, Gönner F, Vogtmann T, Priem R, Fahlbusch, R. Cerebral Hemodynamics in Subarachnoid Hemorrhage Evaluated by Transcranial Doppler Sonography. Part 1. Reliability of Flow Velocities in Clinical Management. Neurosurgery, 1993; 33: 1-9 [ Links ]
42. Heistad, DD, Kontos HA. Cerebral circulation. Handbook of Physiology, "The cardiovascular system III", chpt. 5, 137-82, 1983. [ Links ]
43. Smith RS, Holaday HR, Wilson RG. Transcranial Doppler ultrasonography in neurosurgical practice. Clin Neurosurg, 1989; 37: 255-74. [ Links ]
44. Astrup J. Cerebrovascular physiology. En: Carter LP, Spetzler RF, Hamilton MG (eds) Neurovascular surgery. Mc Graw- Hill, New York, 1995. [ Links ]
45. Chicoine MR, Dacey RG Jr. Clinical aspects of subarachnoid hemorhage. En: Welch KMA, Caplan LR, Reis, DJ, Siesjö BO K, Weir B: Primer on cerebrovascular diseases. Academic press, San Diego, 1997. [ Links ]
46. Takeuchi H, Handa Y, Kobayashi H, Kawano H, Hayashi M. Impairment of cerebral autoregulation during the developement of chronic cerebral vasospasm after subarachnoid hemorhage in primates. Neurosurgery, 1991; 28: 41-8. [ Links ]
47. Yonas H, Sekhar L, Johnson DW, Gur D. Determination of irreversible ischemia by xenon-enhanced computed tomographic monitoring of cerebral blood flow in patients with symptomatic vasospasm. Neurosurgery, 1989; 24: 368-72. [ Links ]
48. Fontana H, Belziti H, Requejo F, Buratti S, Recchia M. La circulación cerebral en condiciones normales y patológicas. Parte II. Las arterias de la base. Rev Arg Neuroc, 2007; 21: 65-70. [ Links ]
49. Newell, D W; Grady, M S; Eskridge, J M; Winn, H R. Distribution of angiographic vasospasm after subarachnoid hemorrhage: implications for diagnosis by transcranial Doppler ultrasonography. Neurosurgery, 1990; 83(3): 394-402. [ Links ]
50. Matsuzaki T, Mizukami M, Kawase T, Tazawa T. Angiographic vasospasm and regional cerebral blood flow in the cases of ruptured aneurysm -Correlation between vasospasm in the middle cerebral artery and delayed ischemic hemiparesis (Jpn). No Shinkei Geka, 1981; 13:1511-6. [ Links ]
51. Grubb RL, Raichle ME Eichling JO, Gado MH. Effects of subarachnoid hemorrhage on cerebral blood volume, blood flow, and oxygen utilization in humans J Neurosurg, 1977;46: 446-53. [ Links ]
52. Tran Dinh YR, Lot G, Benrabah R, Baroudy O, Cophignon J, Seylaz J. Abnormal cerebral vasodilation in aneurysmal hemorrhage: use of serial 133Xe cerebral blood flow measurement plus acetazolamide to assess vasospasm. J Neurosurg, 1993; 79: 490-3. [ Links ]
53. Minhas PS, Menon DK, Smielevski P, Czosnika M, Kirkpatrick PJ, Clark JC, Pckard JD. Positron emission tomographic disturbances and transcranial Doppler findings among patients with neurological deterioration after subarachnoid hemorrhage. Neurosurgery, 2003; 52: 1017-24. [ Links ]
54. Clyde BL, Resnik DK, Yonas H, Smith HA, Kaufmann AM. The relatioship of blood velocity as measured by transcranial Doppler ultrasonography to cerebral blood flow as determined by stable xenon computed tomographic studies after aneurysmal subarachnoid hemorrhage. Neurosurgery, 1996; 10: 896-905. [ Links ]
55. Celsis P, Chan M, Marc-Vergnes JP, Leydet P, Viallard G, Charlet JP, Danet B.Measurement of cerebral circulation in man. Eur J Nucl Med, 1985; 10: 426-31. [ Links ]
56. Cronqvist S, Greitz T. Cerebral circulation time and cerebral blood flow. Acta Radiol (Diagn), 1969; 8: 296-304. [ Links ]
57. Okada Y, Shima T, Nishida M, Yamane K, Hatayama T, Yamanaka C, Yoshida A. Comparison of transcranial Doppler investigation of aneurysmal vasospasm with digital subtraction angiographic and clinical findings. Neurosurgery, 1999; 45: 443-50. [ Links ]
58. Udoetuk JD, Stiefel MF, Hurst RW, Weigele JB, LeRoux PD. Admission angiographic cerebral circulation time may predict subsequent angiographic vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery, 2007; 61: 1152-9. [ Links ]
59. Vermeulen M, Lindsay KW, Van Gijn, J. Subarachnoid Hemorrhage. En la colección: Major problems in neurology. WB Saunders Co. London, 1992. [ Links ]
60. Fontana H, Belziti H. Hipertensión endocraneana y traumatismo encéfalocraneano. Revisión conceptual. Rev Arg Neuroc, 1995; 9: 73-83. [ Links ]
61. Rinkel GJE, Feigin VL, Algra A, van Gijn J. Hypervolemia in aneurysmal subarachnoid hemorrhage. En: Hankey GJ Ed. Cochrane Corner. Stroke, 2005; 36: 1104-5. [ Links ]
62. Hayakawa K, Okuno Y, Miyazawa T, Murayama Ch, Fritz-Zieroth B, Shimizu Y. The decrease of intracarotid pressure during carotid angiography. Inv Radiol. 1994; 29 Supp. 2: S96- S98. [ Links ]
63. Sitoh H, Hyakawa K, Nishimura K, Okuno Y, Murayama Ch, Miyazawa T Fritz-Zierth B, Shimizu Y. Intracarotid blood pressure changes during contrast medium injection. AJNR, 1996; 17: 51-4. [ Links ]
64. Nornes H, Sorteberg W, Nakstad P, Bakke SJ, Aaslid R, Lindegaard, KF. Hemodynamic aspects of clinical cerebral angiography. Acta Neurochir. 1990; 105: 89-97. [ Links ]
65. Yoon DY, Choi CS, Kim KH, Cho B-M. Multidetector-row CT angiography of cerebral vasospasm after aneurysmal subarachnoid hemorrhage: comparison of volume-rendered images and digital subtraction angiography. AJNR, 2006; 27: 370-7. [ Links ]
66. Shimoda M, Oda S, Tsugane R, Sato O. Intracranial complications of hypervolemic therapy in patients with a delayed ischemic deficit attributed to vasospasm. J Neurosurg, 1993; 78: 423-9. [ Links ]
67. Fontana H, Belziti H, Requejo F, Recchia M, Buratti S. La circulación cerebral en condiciones normales y patológicas. Parte I: hipertensión endocraneana e isquemia secundaria. Rev Arg Neuroc, 2007; 21: 33-47. [ Links ]
68. Treggiari-Venzi MM, Suter PM, Romand J-A. Review of medical prevention of vasospasm after aneurysmal subarachnoid hemorrhage : a problem of neurointensive care. Neurosurgery, 2001; 48: 249-62. [ Links ]
69. Soehle M, Czosnika M, Pickard JD, Kirkpatrick PJ. Continuous assessment of cerebral autorregulation in subarachnoid hemorrhage. Anesth Analg, 2004; 98: 1133-9. [ Links ]
70. Lam JMK, Smielevski P, Czosnika M, Pickard JD, Kirkpatrick PJ. Predicting delayed ischemic deficits after aneurysmal subarachnoid hemorrhage using a transient hyperemic response test of cerebral autorregulation. Neurosurgery, 2000; 47: 819-26. [ Links ]
71. Romner B, Brandt L, Berntman L, Algotsson L, Ljunggren B, Messeter K. Simultaneous transcranial Doppler sonography and cerebral blood flow measurements of cerebrovascular CO2-reactivity in patients with aneurysmal subarachnoid haemorrhage. Br J Neurosurg, 1991; 5: 31-7. [ Links ]
72. Frontera JA, Rundek T, Schmidt JM, Claassen J, Parra A, Wartenberg KE, et al. Cerebrovascular reactivity and vasospasm after subarachnoid hemorrhage : a pilot study. Neurology, 2006; 66: 727-9 [ Links ]
73. Marks MP, Steinberg GK, Lane B. Intraarterial papaverine for the treatement of vasospasm. Am J Neuroradiol, 1993; 14: 822-6. [ Links ]
74. Polin RS, Hansen CA, German P, Chadduk JB, Kassell NF. Intra arterially administered papaverine for the treatment of symptomatic cerebral vasospasm. Neurosurgery, 1998; 42: 1256-67. [ Links ]
75. Hoh, BL, Ogilvy CS. Endovascular tretment of cerebral vasospasm: transluminal balloon angioplasty, intra-arterial papaverine and intra-arterial nicardipine. Neurosurgery Clin NA, 2005; 16: 501- 16. [ Links ]

