










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de cardiología
versión On-line ISSN 1850-3748
Rev. argent. cardiol. v.73 n.6 Buenos Aires nov./dic. 2005
ARTICULOS DE REVISIÓN
Las "respuestas lentas" a la angiotensina II, la activación de la proteína Src y del factor de crecimiento epidérmico en la génesis de la hipertensión esencial
Sabas Iván Gómez1, Ezequel A. Piccione2, Carlos A. Feldstein3, J. Carlos Romero4
Este trabajo fue subsidiado por los Institutos Nacionales de la Salud (NIH) de los Estados Unidos de Norteamérica, Grant L I - 16496, y por la Mayo Foundation
1 Becario de la Mayo Clinic Foundation. Departament of Physiology and Biophysics, Rochester, MN
2 Estudiante de Medicina, UBA. Becario, Programa de Pasantías de Mayo Clinic Foundation, Rochester, MN
3 Profesor de Medicina Interna, Jefe del Programa Hipertensión Arterial, Hospital de Clínicas José de San Martín, Facultad de Medicina, UBA
4 Profesor de Fisiología y Medicina Interna. Mayo School of Medicine, Rochester, MN
Recibido: 12/08/2005
Aceptado: 13/09/2005
Dirección para separatas: Prof. J. Carlos Romero, MD Mayo College of Medicine 200 First Street . South West Rochester, Minnesota 55905 USA
e-mail: romero.juan@mayo.edu
RESUMEN
La hipertensión esencial es inducida por disfunción renal. Receptores normotensos de riñones de hipertensos desarrollan hipertensión y viceversa. La alteración renal más importante es el desacople del SRA respecto del nivel de sodio. El estrés oxidativo (ST-OX) es estimulado cuando los niveles de angiotensina II (Ang II) son inapropiados respecto del sodio corporal total. El ST-OX potencia el efecto vasoconstrictor de la Ang II por disminución del óxido nítrico (NO) y/o por incremento de los vasoconstrictores, como isoprostanos, ET1 y otros. Estos efectos se ponen de manifiesto en la "respuesta lenta a la Ang II" en la que la infusión en dosis pequeñas (subpresoras) induce retención de sodio y consecuente estímulo del STOX, con vasoconstricción. Estos efectos están mediados por señales intracelulares como la activación de proteína Src y del receptor del factor de crecimiento epidérmico por la Ang II, que parecen ser un mecanismo de vasoconstricción importante. Las especies reactivas de oxígeno inducidas por estos factores sostendrían una reacción autocatalítica, responsable de la producción sostenida de vasoconstrictores, con lo que se perpetúa la hipertensión.
Palabras clave: Riñón; Trasplante; Sistema renina-angiotensina; Sal; Respuestas presoras; Estrés oxidativo; Señalización intracelular
SUMMARY
Factors Contributing to Essential Hypertension Genesis
Renal alterations related to hypertension are so intrinsic to the kidney that they can be transplanted with the organ. Normotensive recipients that receive a kidney transplant from a hypertensive offspring donor become hypertensive and viceversa. Oxidative stress (OX-ST) is stimulated when plasma levels of angiotensin II (Ang II) become inappropriate compared to total body sodium or viceversa. OX-ST potentiates the vasoconstrictor effects of Ang II decreasing nitric oxide, and/or stimulating vasoconstrictors such as isoprostanes, endothelins, etc. These effects are present in the so called "slow responses" to Ang II, where the prolonged infusion of sub-pressor doses of Ang II induces sodium retention and OX-ST, sensitizing the organism to vasoconstriction. These effects are mediated by a set of intracellular signals, the most important of which seems to be the Ang II induced activation of Src protein and epidermal growth factor. The production of superoxide induced by these factors could be sustained by an auto-catalytic reaction that accounts for vasoconstriction.
Key words: Kidney; Transplant; Renin-angiotensin system; Salt; Pressor esponses; Oxidative stress; Intacellular signaling
ABREVIATURAS
Ang II Angiotensina II
ARP Aumento de renina plasmática
ET1 Endotelina 1
FCE-LH Factor de crecimiento epidérmico ligado a heparina
IECA Inhibidores de la enzima convertidora
NADPH Forma reducida del fosfato de dinucleótido de nicotinamida
y adenina
NO Óxido nítrico
PA Presión arterial
PKC Proteinquinasa C
RFCE Receptor del factor de crecimiento epidérmico
SRA Sistema renina-angiotensina
ST-OX Estrés oxidativo
INTRODUCCIÓN
En la caracterización de la patogenia de la hipertensión esencial se han logrado adelantos significativos, los cuales representan aportes importantes para establecer los criterios diagnósticos, la elección del tratamiento y efectuar el seguimiento del paciente. En el presente trabajo se efectúa una revisión de estos conocimientos recientes y se plantean hipótesis que ponen énfasis sobre la importancia de la interrelación de esos factores causales.
EL RIÑÓN COMO ÓRGANO GENERADOR DE HIPERTENSIÓN
Hipertensión y trasplante renal
El papel de la disfunción renal en la instalación de la hipertensión arterial se pone de manifiesto de manera ostensible durante los trasplantes renales. (1-8) En los trasplantes cruzados entre cepas de ratas hipertensas y normotensas, los animales receptores fueron nefrectomizados para evitar cualquier confusión por el efecto de los riñones nativos. La presión arterial (PA) del receptor cambió después del procedimiento y alcanzó niveles similares a los del donante. (5-8) Dahl y colaboradores (1, 2) trasplantaron riñones de ratas hipertensas sensibles a la sal en ratas normotensas resistentes a la sal y comprobaron que los animales receptores desarrollaron hipertensión. Por el contrario, cuando los riñones de ratas resistentes a la sal se trasplantaron en ratas sensibles a la sal, estas últimas no desarrollaron hipertensión. (2, 3) El trasplante renal de ratas jóvenes normotensas resistentes a la sal en ratas adultas hipertensas sensibles a la sal indujo en éstas un descenso significativo de la PA. El factor determinante de la sensibilidad a la sal en ratas sería un desplazamiento hacia la derecha de la curva presión-natriuresis. Es decir, la cepa sensible a la sal requiere una presión de perfusión renal mayor que la cepa resistente a la sal para excretar la misma cantidad de sodio. (4, 9) Otros investigadores (10) sugirieron que la sensibilidad a la sal se debería a una adaptación anormal de la resistencia periférica a la expansión de volumen. Bianchi y colaboradores (11) observaron el desarrollo de hipertensión en ratas nefrectomizadas de una cepa normotensa al trasplantar en ellas riñones de ratas jóvenes de una cepa hipertensa (pero todavía normotensas a esa edad). También demostraron que el trasplante renal de ratas adultas normotensas en las hipertensas indujo un descenso sostenido de la PA. (5) Rettig y colaboradores (6, 7) administraron inhibidores de la enzima convertidora (IECA) para normalizar la presión de perfusión renal en ratas espontáneamente hipertensas. Luego efectuaron el trasplante de un riñón de rata hipertensa tratada en otra normotensa nefrectomizada, que en ésta última indujo hipertensión. Esto demostró que una alteración renal "silente" se expresa tarde o temprano, incluso luego de un período de normalización tanto de la PA como de la presión de perfusión renal alcanzado por medio de tratamiento farmacológico.
Otras evidencias del papel primario del riñón en la patogénesis de la hipertensión surgieron de experiencias efectuadas en seres humanos. Los receptores de un trasplante renal desarrollaron hipertensión no relacionada con la PA pretrasplante o con la nefropatía que motivó el trasplante. (8) Guidi y colaboradores (12) comprobaron en receptores de riñones cadavéricos que aquellos sin antecedentes familiares de hipertensión requirieron más terapia antihipertensiva cuando el donante pertenecía a una familia con antecedentes de hipertensión. Esto sugiere la transmisión de hipertensión desde un donante de familia hipertensa a un receptor de familia normotensa. Los pacientes con nefroesclerosis secundaria a hipertensión que luego de ser nefrectomizados recibieron riñones de donantes jóvenes normotensos normalizaron su PA. (13) Después de un seguimiento promedio de 4 años (máximo 8 años), la PA se mantenía normal sin medicación antihipertensiva. (13)
EL PAPEL DEL SISTEMA RENINA-ANGIOTENSINA
La disfunción renal con activación del sistema reninaangiotensina (SRA) podría ser el proceso subyacente más importante que conduce al desarrollo de la hipertensión arterial. La Ang II posee acciones presoras rápidas y lentas. La "respuesta rápida" a la Ang II se caracteriza por una contracción inmediata del músculo liso arteriolar, que alcanza su máximo en segundos y desaparece en dos a tres minutos. Podría constituir sólo un efecto farmacológico, pues es inducida por dosis altas y los niveles plasmáticos de Ang II necesarios para incrementar la PA son mayores de 2.500 pg/ml. Esta concentración no se alcanza en ninguna situación fisiológica (depleción severa de sodio) o patológica (hipertensión renovascular o hemorragia masiva). La "respuesta lenta" estaría involucrada en la producción de hipertensión experimental y humana. (14) Consiste en una elevación progresiva de la PA por administración continua de dosis subpresoras de Ang II (inferiores al umbral de vasoconstricción aguda). La hipertensión se desarrolla entre 4 y 7 días después de iniciada la infusión. Sólo hay un aumento transitorio de la Ang II plasmática, que luego se mantiene dentro de límites comparables a los previos a la infusión. Esto es muy similar a lo que se comprueba en la hipertensión esencial establecida, que se caracteriza por un aumento aislado de la resistencia periférica.
ACTIVACIÓN DEL ESTRÉS OXIDATIVO
El aumento de PA en la "respuesta lenta" a la Ang II es similar en magnitud al que se alcanza (transitoriamente) en la "respuesta rápida". La Ang II activaría un mecanismo secundario, que induce hipertensión, distinto del que media la vasoconstricción aguda. (14) En la actualidad se reconoce que ese mecanismo consiste en la activación del estrés oxidativo (ST-OX), que produce hipertensión por depleción de los mediadores vasodilatadores e incremento de los vasoconstrictores, cuya consecuencia es el aumento de la resistencia vascular periférica. Haas y colaboradores, (15) en cerdos, y Reckelhoff y colaboradores, (16) en ratas, demostraron un incremento de isoprostanos plasmáticos inducido por la Ang II, que se halla involucrado en el desarrollo de hipertensión. La infusión crónica de Ang II induce la expresión de NADPH oxidasa, que aumenta los niveles de anión superóxido. (17) Éste, a su vez, puede combinarse con el óxido nítrico (NO), lo cual da origen a peroxinitrito (Figura 1). El peroxinitrito posee gran poder oxidativo y actúa no enzimáticamente sobre el ácido araquidónico, para producir mediadores vasoconstrictores como tromboxano A2 e isoprostanos. La depleción de NO produce vasoconstricción e instalación de hipertensión. Cuando se administró un inhibidor selectivo de la síntesis de NO en ratas, el efecto vasoconstrictor de la Ang II se potenció. (18) El peroxinitrito también depleciona la prostaciclina por inhibición de la prostaciclina sintetasa (Figura 2). La Ang II tiene efectos directos sobre los prostanoides al activar la lipooxigenasa, con disminución de la conversión del endoperóxido H2 (vasoconstrictor) en prostaciclina (vasodilatador). (19) La endotelina-1 (ET1) podría intervenir en las respuestas lentas mediadas por la Ang II. Sería estimulada no sólo en forma directa por la Ang II, sino también indirectamente por los isoprostanos. (15, 16, 20, 22)
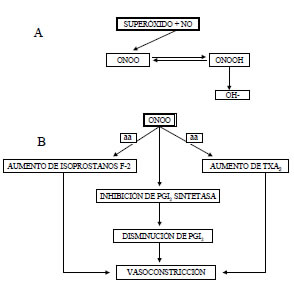
Fig. 1. A. La combinación de NO y superóxido produce peroxinitrito (ONOO), que se halla en equilibrio reversible con ácido peroxinitroso (ONOOH). Éste es una fuente de radical oxidrilo (OH-). B. El ONOO produce vasoconstricción al convertir el ácido araquidónico (aa) en isoprostanos F-2 y por incremento del tromboxano A2. El ONOO también causa vasoconstricción al inhibir la prostaciclina (PGI2) sintetasa.
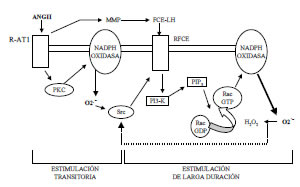
Fig. 2. Modelo propuesto para la generación de especies reactivas de oxígeno (ERO) por Ang II. Ésta se liga al receptor específico AT1 con una generación rápida de ERO a través de la activación de la PKC. Las ERO activan la Src con la consecuente transactivación del receptor de factor de crecimiento epidérmico (RFCE). La proteína fosfatidil inositol 3-quinasa (PI3-K) produce fosfatidilinositol 3-fosfato (PIP3) que activa Rac, proteínas de la membrana celular acopladoras de GTP. La Rac-GTP se une al complejo de la NADPH oxidasa y lleva a una mayor generación de ERO, que estimulan a la Src. Esta última amplifica la actividad de la NADPH oxidasa. La transactivación del RFCE también se efectúa vía extracelular. Esto involucra la activación de metaloproteinasas que liberan factor de crecimiento epidérmico ligado a heparina (FCE-LH) al medio extracelular. Finalmente, se produce la interacción entre el FCE-LH con el RFCE. ONOO: peroxinitrito. ONOOH: ácido peroxinitroso. OH-: radical oxhidrilo. aa: ácido araquidónico. PGI2: prostaciclina. R-AT1: receptor AT1 de Ang II. PKC: proteinquinasa C. PI3-K: fosfatidilinositol 3-quinasa. PIP3: fosfatidil inositol 3-fosfato. MMP: metaloproteinasas. RFCE: receptor del factor de crecimiento epidérmico. Rac: sustrato de toxina botulínica C3 ras-relacionado. GDP: guanosina difosfato: GTP: guanosina trifosfato. FCE-LH: factor de crecimiento epidérmico ligado a heparina. O2 -= anión superóxido.H2O2: peróxido de hidrógeno.
PAPEL DEL DESACOPLE ENTRE SODIO Y ANG II
La retención inapropiada de sodio parece tener un papel preponderante en la producción y el mantenimiento de las "respuestas lentas" a la Ang II. La depleción de sodio se acompaña de contracción del compartimiento extracelular que estimula el aumento de los niveles de renina plasmática (ARP) y Ang II. En situaciones patológicas como la hipertensión por estenosis de la arteria renal o la hipertensión esencial, los niveles aumentados o normales de Ang II coexisten con volumen extracelular normal o expandido, respectivamente, lo cual constituye una disociación de la relación normal entre estos parámetros. Hollenberg (23) introdujo el concepto de "niveles excesivos" de Ang II respecto de los de sodio. Observó que un grupo de pacientes hipertensos no reducía sus niveles de ARP durante la expansión de volumen. Por esta razón, los llamó "no moduladores". La infusión de dosis pequeñas de Ang II previno la supresión del ARP provocada por la administración de soluciones salinas. (24) El aumento de PA fue proporcional a la expansión de volumen y resultó mayor que en animales a los que sólo se les administró exceso de sal. Este experimento demostró que si el nivel circulante de Ang II se mantiene constante, los valores de PA estarán determinados por la ingesta de sodio. En esas condiciones, la administración de furosemida produjo normalización de la PA; esto sugiere que la reducción de volumen provocada por el diurético hizo que los niveles de Ang II se volviesen "apropiados" para el volumen extracelular. Esto podría explicar el beneficio terapéutico de los diuréticos en la hipertensión. También explicaría la acción antihipertensiva de los IECA, que disminuyen los niveles de Ang II haciéndolos "apropiados" a los de sodio. Sánchez y colaboradores (25) observaron que una dieta hiposódica prolongada se acompañó de niveles de ARP más elevados y de una actividad menor de calicreínas urinarias en los hipertensos no moduladores que en los moduladores. Pareciera que la actividad menor del sistema calicreína está asociada con la falla en el manejo del sodio. Hay suficientes evidencias que permiten postular que en la hipertensión esencial existiría un desacople o disociación de la relación entre Ang II y sodio. La Ang II mantendría la hipertensión a través de la activación del estrés oxidativo. Murphey y colaboradores (26) midieron la activación de estrés oxidativo luego de administrar Ang II en pacientes hipertensos y normotensos puestos sucesivamente bajo dietas hipersódica e hiposódica. La Ang II sólo produjo un incremento del estrés oxidativo (expresado por aumento de isoprostanos) en los hipertensos con dieta hipersódica. Este estudio fue el primero en demostrar directamente que la Ang II aumenta los metabolitos responsables del estrés oxidativo en seres humanos hipertensos.
PAPEL DE LOS MECANISMOS INTRACELULARES
Los efectos del SRA se expresan de manera característica en la señalización intracelular, cuyo análisis resulta importante porque explica los rasgos adaptativos del organismo y muestra posibles blancos terapéuticos para ser tenidos en cuenta para influir selectivamente diferentes formas de hipertensión.
Respuesta rápida a la Ang II: en ésta, la Ang II se liga al receptor específico tipo AT1 acoplado a proteína G, que activa la fosfolipasa C. Ésta induce el clivaje del fosfatidilinositol produciendo inositol 3- fosfato, responsable de la liberación de calcio desde el retículo endoplasmático, lo cual genera vasoconstricción. Esta cadena de reacciones tradicionalmente se ha aceptado como responsable de la contracción del músculo liso vascular estimulada por la Ang II.
Respuesta lenta a la Ang II: como el calcio proveniente del retículo endoplasmático no es suficiente para sostener la respuesta vasoconstrictora, se sugirió que la apertura de los canales del calcio de la membrana celular con ingreso del catión desde el espacio extracelular mantenía la contracción del músculo liso arteriolar. Este concepto se modificó con el descubrimiento del estrés oxidativo y su participación en la regulación de la PA. Estudios posteriores identificaron el papel de la proteinquinasa C (PKC) que durante la estimulación por la Ang II es activada por calcio y diacilglicerol. La PKC estimula la NADPH-oxidasa, responsable de la liberación de anión superóxido.
¿El estrés oxidativo está involucrado en la respuesta lenta a la Ang II?: la incubación prolongada de Ang II con músculo liso arterial produjo un aumento rápido del anión superóxido, mediado parcialmente por la PKC. (27) Después de alcanzar un pico a los treinta segundos, el nivel del anión superóxido descendió hasta casi los niveles de control y luego aumentó en forma progresiva para alcanzar una meseta a los treinta minutos. Al inhibir la PKC, se bloqueó completamente el incremento inicial, pero sólo se redujo en un 50% la elevación tardía, lo cual sugiere la participación de otros factores. Estudios posteriores mostraron que la Ang II activaría el receptor del factor de crecimiento epidérmico (RFCE) a través de una proteinquinasa de tirosina llamada Src (Figura 2). Esto sería seguido por la activación de fosfatidilinositol 3-quinasa (PI3-K) y de proteína G monomérica (Rac GTP), proceso que culmina con la estimulación de la NADPH oxidasa y producción de anión superóxido. Éste tiene una participación especial, porque retroestimularía la Src que activa aún más el RFCE. Estas reacciones características son compatibles con el desarrollo de las respuestas lentas a la Ang II, ya que en forma prolongada se produce un efecto amplificado por un sistema de retroalimentación. Explicando este fenómeno en forma diferente, la administración de dosis subpresoras de Ang II requiere un período de latencia durante el cual los mecanismos intracelulares de la respuesta aguda (vía fosfatidilinositol) no están involucrados, pero sí lo está el sistema Src-RFCE que progresivamente aumentará la formación del anión superóxido. De aquí en adelante, la estimulación continua del estrés oxidativo potenciará y sostendrá los efectos vasoconstrictores de la Ang II a través de la depleción de NO, de la síntesis de isoprostanos y otros mecanismos.
Papel de otros factores intracelulares en la hipertensión: estudios recientes mostraron que la inhibición selectiva de la activación del RFCE reduce la vasoconstricción producida por la Ang II y la noradrenalina. (28) Es decir, el RFCE se relacionó con vasoconstricción y consecuentemente con el desarrollo de hipertensión. El mecanismo propuesto involucra una vía extracelular que finaliza en la transactivación del RFCE. La interacción de vasoconstrictores (Ang II, ET1 o catecolaminas) con sus receptores acoplados a proteína G activaría las metaloproteinasas. Estas últimas liberarían el factor de crecimiento epidérmico ligado a heparina (FCE-LH) hacia el medio extracelular, donde mediante la activación de su receptor específico (RFCE) produciría vasoconstricción. Los factores que contribuyen a la activación del RFCE tendrían un papel importante en los estadios tempranos de la hipertensión. Así, se comprobó en ratas espontáneamente hipertensas que el RFCE se activa desde la quinta hasta la duodécima semana. (29) La activación decrece después de transcurrido ese período. El papel del RFCE en la fisiopatología de la hipertensión esencial no se limitaría a su efecto vasoconstrictor. Es bien conocido el papel de la Ang II en la cardiopatía y en la vasculopatía que complican la hipertensión arterial. (27, 28) Se ha sugerido que el RFCE es uno de los mediadores en la estimulación de la hipertrofia y la hiperplasia de los cardiomiocitos y del músculo liso vascular. (28, 29) Aunque este mecanismo recién se está empezando a estudiar, los bloqueantes de la transactivación del RFCE (inhibidores de las metaloproteinasas) podrían tener efectos antihipertensivos al inhibir tanto la vasoconstricción patológica como la proliferación celular.
CONCLUSIONES
El desarrollo de hipertensión esencial subyace en una disfunción renal en la que se altera el acople entre el SRA y el equilibrio hidroelectrolítico. Tal disfunción parece ser intrínseca del riñón, pues se puede transferir con el trasplante del órgano. El desacople del SRA ocurre cuando los niveles circulantes de Ang II son excesivos o inapropiados para la concentración de sodio o el volumen de líquidos corporales. El mecanismo intrínseco responsable es difícil de precisar; podría deberse a la falla en alguno de los múltiples procesos que permiten que el riñón ajuste los niveles de Ang II a los de sodio. Este fenómeno es reproducido por la llamada "respuesta lenta" a la Ang II durante la infusión de dosis muy pequeñas (subpresoras) de ésta que llevan a la retención de sodio. En estas condiciones, la alteración se puede corregir ya sea inhibiendo los efectos de la Ang II (antagonistas de Ang II, IECA), con la administración de diuréticos o por la combinación de ambos tipos de fármacos. El desacople entre la Ang II y el volumen de líquidos corporales estimula el estrés oxidativo, que potencia el efecto vasopresor de la Ang II al reducir la concentración de NO y aumentar la de compuestos vasoconstrictores como la ET1 y los isoprostanos.
Mencionamos otros efectos muy importantes de la Ang II, como la estimulación de la síntesis de colágeno, la hipertrofia celular y la inflamación. En estos procesos, los niveles de Ang II son relevantes sólo cuando se consideran en relación con la carga de sodio. Están involucrados mecanismos intracelulares, de los que examinamos solamente los que participan en las respuestas rápida y lenta. Se ha puesto énfasis en la participación de la proteína Src y del RFCE. La activación de estos últimos mediadores podría, en teoría, generar hipertensión, pues son efectores de varios vasoconstrictores. Además, podrían constituir potenciales blancos para el desarrollo de nuevas clases de fármacos antihipertensivos.
1. Dahl LK, Heine M, Thompson K. Genetic influence of renal homografts on the blood pressure of rats from different strains. Proc Soc Exp Biol Med 1972;140:852-6. [ Links ]
2. Dahl LK, Heine M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ Res 1975;36:692-6. [ Links ]
3. Dahl LK, Heine M, Thompson K. Genetic influence of the kidneys on blood pressure. Evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ Res 1974; 40:94-101. [ Links ]
4. Maude DL, Kao-Lo G. Salt excretion and vascular resistance of perfused kidneys of Dahl rats. Hypertension 1982;4:532-7. [ Links ]
5. Bianchi G, Fox U, Di Francesco GF, Giovanetti AM, Pagetti D. Blood pressure changes produced by kidney cross-transplantation between spontaneously hypertensive rats and normotensive rats. Clin Sci Mol Med 1974;47:435-48. [ Links ]
6. Rettig R, Stauss H, Folberth C, Ganten D, Waldherr B, Unger T. Hypertension transmitted by kidneys from stroke-prone spontaneously hypertensive rats. Am J Physiol 1989;257:F197-203. [ Links ]
7. Rettig R, Folberth C, Stauss H, Kopf D, Waldherr R, Unger T. Role of the kidney in primary hypertension: a renal transplantation study in rats. Am J Physiol 1990;258:F606-11. [ Links ]
8. Merino GE, Kjellstrand CM, Simmons RL, Najarian JS. Late hypertension in renal transplant recipients: possible role of the donor in late primary hypertension. Proc Clin Dial Transplant Forum 1976;6:145-52. [ Links ]
9. Girardin E, Caverzasio J, Iwai J, Bonjour JP, Muller AF, Grandchamp A. Pressure natriuresis in isolated kidneys from hypertensionprone and hypertension-resistant rats (Dahl rats). Kidney Int 1980; 18:10-9. [ Links ]
10. Greene AS, Yu ZY, Roman RJ, Cowley AW Jr. Role of blood volume expansion in Dahl rat model of hypertension. Am J Physiol 1990;258:H508-14. [ Links ]
11. Bianchi G, Fox U, Di Francesco GF, Bardi U, Radice M. The hypertensive role of the kidney in spontaneously hypertensive rats. Clin Sci Mol Med Suppl 1973;45:135s-9. [ Links ]
12. Guidi E, Bianchi G, Rivolta E, Ponticelli C, Quarto di Palo F, Minetti L, et al. Hypertension in man with a kidney transplant: role of familial versus other factors. Nephron 1985;41:14-21. [ Links ]
13. Curtis JJ, Luke RG, Dustan HP, Kashgarian M, Whelchel JD, Jones P, et al. Remission of essential hypertension after renal transplantation. N Engl J Med 1983;309:1009-15. [ Links ]
14. Dickinson CJ, Lawrence JR. A slowly developing pressor response to small concentrations of angiotensin. Its bearing on the pathogenesis of chronic renal hypertension. Lancet 1963;1:1354-6. [ Links ]
15. Haas JA, Krier JD, Bolterman RJ, Juncos LA, Romero JC. Lowdose angiotensin II increases free isoprostane levels in plasma. Hypertension 1999;34:983-6. [ Links ]
16. Reckelhoff JF, Zhang H, Srivastava K, Roberts LJ 2nd, Morrow JD, Romero JC. Subpressor doses of angiotensin II increase plasma F(2)-isoprostanes in rats. Hypertension 2000;35:476-9. [ Links ]
17. Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res 2002;90:E58-65. [ Links ]
18. Kitamoto S, Egashira K, Kataoka C, Usui M, Koyanagi M, Takemoto M, et al. Chronic inhibition of nitric oxide synthesis in rats increases aortic superoxide anion production via the action of angiotensin II. J Hypertens 2000;18:1795-800. [ Links ]
19. Lin L, Balazy M, Pagano PJ, Nasjletti A. Expression of prostaglandin H2-mediated mechanism of vascular contraction in hypertensive rats. Relation to lipoxygenase and prostacyclin synthase activities. Circ Res 1994;74:197-205. [ Links ]
20. Alexander BT, Cockrell KL, Rinewalt AN, Herrington JN, Granger JP. Enhanced renal expression of preproendothelin mRNA during chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol 2001;280:R1388-92. [ Links ]
21. Fukunaga M, Yura T, Badr KF. Stimulatory effect of 8-Epi-PGF2 alpha, an F2-isoprostane, on endothelin-1 release. J Cardiovasc Pharmacol 1995;26:S51-2. [ Links ]
22. Ruef J, Moser M, Kubler W, Bode C. Induction of endothelin-1 expression by oxidative stress in vascular smooth muscle cells. Cardiovasc Pathol 2001;10:311-5. [ Links ]
23. Hollenberg NK, Chenitz WR, Adams DF, Williams GH. Reciprocal influence of salt intake on adrenal glomerulosa and renal vascular responses to angiotensin II in normal man. J Clin Invest 1974; 54:34-42. [ Links ]
24. DeClue JW, Guyton AC, Cowley AW Jr, Coleman TG, Norman RA Jr, McCaa RE. Subpressor angiotensin infusion, renal sodium handling, and salt-induced hypertension in the dog. Circ Res 1978;43:503-12. [ Links ]
25. Sanchez R, Gimenez MI, Ramos F, Baglivo H, Ramirez AJ. Nonmodulating hypertension: evidence for the involvement of kallikrein/ kinin activity associated with overactivity of the renin-angiotensin system. Successful blood pressure control during long-term Na+ restriction. J Hypertens 1996;14:1287-91. [ Links ]
26. Murphey LJ, Morrow JD, Sawathiparnich P, Williams GH, Vaughan DE, Brown NJ. Acute angiotensin II increases plasma F2- isoprostanes in salt-replete human hypertensives. Free Radic Biol Med 2003;35:711-8. [ Links ]
27. Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 2002;91:406-13. [ Links ]
28. Hao L, Du M, Lopez-Campistrous A, Fernandez-Patron C. Agonistinduced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ Res 2004;94:68-76. [ Links ]
29. Ferder L, Romano LA, Ercole LB, Stella I, Inserra F. Biomolecular changes in the aging myocardium: the effect of enalapril. Am J Hypertens 1998;11:1297-304. [ Links ]

