










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de dermatología
versión On-line ISSN 1851-300X
Rev. argent. dermatol. v.87 n.4 Ciudad Autónoma de Buenos Aires oct./dic. 2006
Porfirias poco frecuentes. Casos detectados en la población argentina
Non frequent porphyrias in the argentinean population
V A Melito*, M V Rossetti**, V E Parera*** y A Batlle****
* Ayudante de Primera - FCEyN - UBA, Profesional Principal - CONICET
** Profesora Adjunta - FCEyN - UBA, Investigadora Independiente - CONICET, Vicedirectora 1ra del CIPYP
*** Profesora Adjunta - FCEyN - UBA, Investigadora Independiente - CONICET, Vicedirectora 2da del CIPYP
**** Investigadora Superior - CONICET, Directora del CIPYP
Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP) - CONICET - UBA - Hospital de Clínicas José de San Martín.
e-mail: batlle@mail.retina.ar; batlle@fibertel.com.ar
Fecha de recepción: 30.08.06
Fecha de aceptación: 29.09.06
RESUMEN: Las porfirias son consecuencia de fallas en el metabolismo del hemo. Se clasifican según el tipo de sintomatología clínica prevalente o el órgano donde se expresa preferencialmente la falla metabólica. En general la deficiencia enzimática está asociada a mutaciones en los genes que codifican para cada una de las enzimas.
Están descritos 7 tipos de porfiria diferentes. Se transmiten por carácter autosómico dominante a excepción de la PCE, la PHE y la NPA que son recesivas. Sin embargo, están reportadas variantes homocigotas para el resto de las porfirias de pronóstico y evolución mucho más grave que la forma heterocigota.
La descripción de estos casos poco frecuentes, sus tratamientos y evolución, facilitarían tanto el diagnóstico diferencial de la porfiria como el conocimiento de las posibilidades terapéuticas en cada caso.
Asimismo para las porfirias heterocigotas con manifestación infantil, su identificación temprana y tratamiento aseguraría una mejor evolución minimizando los riesgos asociados.
Se han diagnosticado 5 casos de porfirias agudas en niñas: 2 de PAI, 2 de PV y 1 de CPH. Entre las porfirias cutáneas se presentan 25 casos de PCT infantil, el primer caso de PHE en Argentina, 4 casos de PCE infantil y 1 en un adulto y 2 casos de PPE con compromiso hepatobiliar.
PALABRAS CLAVE: Enzimas; Hemo; Porfirias; Porfirias agudas; Porfirias cutáneas.
SUMMARY: The Porphyrias are a group of diseases resulting from partial deficiencies in one of the heme biosynthetic enzymes. These disorders can be classified on the basis of their clinical manifestations or according the organ where the metabolic deficiency is mainly expressed. In general this enzyme deficiency is associated with mutations in the genes which codify each enzyme.
There are 7 types of Porphyrias. They are autosomal dominant disorders with the exception of PCE, PHE and NPA which are recessive. However, some rare and severe cases with recessive inheritance have also been reported.
The description of these infrequent cases and their treatments and evolution would make easier the differential diagnosis of Porphyrias as well as the therapeutic possibilities to be applied in each case.
Moreover, it is very important the early identification and treatment of infantile heterozygous porphyrias to avoid the risks of associatedd complications.
In the CIPYP we have diagnosed 5 cases of infantil Acute Porphyrias: 2 PAI, 2 PV and 1 CPH. In the group of Cutaneous Porphyrias we present 25 cases of infantil PCT, the first case of PHE in Argentina, 4 cases of infantil PCE and 1 adult PCE and 2 cases of PPE with hepatic failure.
KEY WORDS: Enzymes; Heam; Porphyrias; Acute porphyrias; Cutaneous porphyries.
INTRODUCCIÓN
Las porfirias son enfermedades metabólicas, hereditarias o adquiridas, producidas como consecuencia de fallas enzimáticas en el camino metabólico del hemo (Fig 1). Esta falla específica, parcial primaria, causa la sobreproducción y acumulación del sustrato de la enzima afectada con un patrón típico dando lugar a una sintomatología clínica característica para cada porfiria que puede incluir cuadros de variada fotosensibilidad cutánea y/o síndrome neuroabdominal 1.

Fig 1: Biosíntesis del Hemo: enzimas involucradas y porfirias asociadas.
Se han descrito 7 tipos diferentes de porfirias que se pueden clasificar en hepáticas o eritropoyéticas, según el órgano en el cual se acumulan principalmente los intermediarios del metabolismo del hemo, o en agudas y/o cutáneas según la sintomatología clínica asociada. Recientemente se ha propuesto una clasificación que unifica ambos criterios que brinda una descripción integral para cada tipo de porfiria (Fig 2). La prevalencia de las porfirias varía desde 0,5 a 10 por 100.000 habitantes en diferentes poblaciones 2.
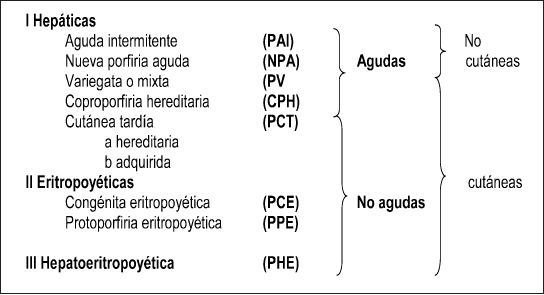
Fig 2: Clasificación de las Porfirias.
Las deficiencias enzimáticas responsables de estas patologías están asociadas generalmente a mutaciones en el gen que codifica para cada enzima. Estas alteraciones genéticas se heredan principalmente según un carácter autosómico dominante. Pertenecen a este grupo la porfiria aguda intermitente (PAI), la porfiria variegata (PV), la coproporfiria hereditaria (CPH), la porfiria cutánea tardía (PCT) y la protoporfiria eritropoyética (PPE). Si bien en estas porfirias la actividad de la enzima afectada es aproximadamente el 50% del valor normal, indicando que el alelo mutado asociado presenta escasa o nula actividad, ésta es suficiente para suplir la demanda celular de hemo. Esto explicaría una penetrancia clínica de sólo alrededor del 20% entre los individuos portadores de la mutación, quienes llegan a manifestar la enfermedad como consecuencia de la exposición a factores desencadenantes. La elevada frecuencia de estas mutaciones en la población general sería la razón de la existencia de las variantes homocigotas descriptas 3.
Las porfirias que se heredan en forma autosómica recesiva son la porfiria congénita eritropoyética (PCE), la nueva porfiria aguda (NPA) y la porfiria hepatoeritropoyética (PHE).
PORFIRIAS AGUDAS
Dentro de las denominadas porfirias agudas se incluyen la PAI, la PV, la CPH y la NPA. Clínicamente se caracterizan por la aparición, generalmente después de la pubertad, de ataques agudos indistinguibles en todos los casos. Son más frecuentes en mujeres. Durante el ataque agudo se encuentran síntomas muy variados que van desde ansiedad, cambios de comportamiento, cansancio y desgano, dolores difusos en extremidades y espalda, hipertensión y taquicardia, hasta el síndrome neuroabdominal con dolores agudos, vómitos y constipación. El deterioro del sistema nervioso central (SNC) puede llevar a la manifestación de convulsiones y confusión, mientras que el progreso de la neuropatía sensomotora puede conducir a una parálisis respiratoria. Como consecuencia de la neuropatía pueden presentarse arritmias que preceden a un mal funcionamiento cardíaco. En los cuadros graves la causa más frecuente de muerte es la neumonía asociada a la asistencia respiratoria mecánica prolongada. El deterioro progresivo del SNC junto con la polineuropatía son poco frecuentes en otras patologías, por lo tanto su presencia simultánea debe ser causa de sospecha de un cuadro de porfiria aguda 4.
Las porfirias agudas son hepáticas y entre ellas la PAI y la NPA son no cutáneas. En la PV y la CPH se pueden manifestar también lesiones cutáneas típicas causadas por el fotodaño, provocado por la presencia de la elevada cantidad de porfirinas circulantes, por lo que se las conoce como porfirias mixtas (Fig 2).
Desde el punto de vista bioquímico se encuentra elevado el contenido de los precursores en orina, ácido δ-aminolevúlico (ALA) y porfobilinógeno (PBG), principalmente durante las crisis agudas. La excreción y el patrón de porfirinas urinarias, fecales y plasmáticas también están alterados.
Porfiria aguda intermitente:
En esta porfiria la falla enzimática se encuentra a nivel de la enzima porfobilinógeno deaminasa (PBG-D) o hidroximetilbilano sintetasa (HMB-S) que sintetiza el hidroximetilbilano (HMB).
Se han descrito dos formas enzimáticas, codificadas por un único gen que por un splicing alternativo da lugar a las formas eritroidea y ubicua. En la expresión más común de esta enfermedad la actividad de la enzima está disminuida en todos los tejidos, mientras que en la denominada PAI variante la actividad de la enzima en eritrocitos es normal.
Esta porfiria es genéticamente heterogénea, encontrándose descriptas alrededor de 246 mutaciones distintas en el gen que codifica la PBG-D (Human Gene Mutation Database: http://www.hgmd.org). Tiene una alta incidencia en la población de países escandinavos (1:10.000).
Durante la fase aguda de la enfermedad la característica clínica es el síndrome neuroabdominal, acompañado por convulsiones, neuropatías periféricas, confusión mental entre otros signos comunes a las porfirias agudas, que pueden o no ocurrir simultáneamente. El desencadenamiento de estos episodios responde en general a la ingesta de determinadas drogas, hormonas, alcohol, dieta hipocarbonada y estrés. El contenido de ALA, PBG y porfirinas en orina y porfirinas en plasma se encuentran elevados. Durante la remisión clínica estos valores aún pueden continuar alterados. Los primeros ataques suceden luego de la pubertad, sin embargo se han descripto algunos pocos casos en niños5,6 entre los cuales deben considerarse los extremadamente poco frecuentes casos de PAI en homocigosis o doble heterocigosis, en los que la actividad de la PBG-D se encuentra disminuída hasta el 1-17% del valor normal. En estos pacientes el pronóstico y calidad de vida son muy malos 7,8,9,10,11.
Porfiria variegata:
Es la segunda porfiria en cuanto a su incidencia y es consecuencia de la actividad disminuida de la enzima mitocondrial protoporfirinógeno oxidasa (PPOx). Es otra porfiria genéticamente heterogénea para la que están descritas 130 mutaciones diferentes en el gen que codifica la PPOx. (Human Gene Mutation Database: http://www.hgmd.org).
La PV se conoce también como porfiria sudafricana dado que es la porfiria más común en ese país. Su incidencia es de 4 casos cada 1.000 habitantes y su introducción en la región se debe a una de las primeras familias holandesas que se instalaron en Ciudad del Cabo hace más de 300 años. Se estima que aproximadamente 2000 descendientes de esta familia heredaron la misma mutación (R59W) en el gen de la PPOX.
Las manifestaciones de la PV ocurren también después de la pubertad con una expresión variada de su sintomatología: dolores abdominales, fragilidad cutánea, fotosensibilidad y trastornos neurológicos; combina los síntomas de las porfirias cutáneas y agudas por lo cual también se la conoce como porfiria mixta, aunque no siempre se observan ambos signos simultáneamente. En los individuos sintomáticos se encuentra un aumento de los precursores ALA y PBG en orina durante el ataque, que son los responsables de los síntomas neurológicos. Durante la fase de remisión estos valores están normales. También puede haber un aumento de porfirinas, principalmente protoporfirina, que se excreta por heces y en menor proporción coproporfirina, responsables de la sintomatología cutánea. En plasma, el contenido de porfirinas está siempre elevado. Aproximadamente un 75% de los individuos portadores de la falla genética pueden permanecer asintomáticos aunque con riesgo de desarrollar crisis neuroabdominales si se exponen a factores desencadenantes como hiponutrición, ingesta de drogas porfirinogénicas como barbituratos, sulfonamidas, anestésicos, alcohol, hormonas esteroides y hasta por alteraciones psicológicas como el estrés.
También para esta porfiria se han descripto casos homocigotas 12,13,14,15,16,17,18.
Coproporfiria Hereditaria:
Entre las porfirias heredadas en forma autosómica dominante, la CPH es la de menor incidencia en la población. En este caso la deficiencia enzimática está localizada en la enzima mitocondrial coproporfirinógeno oxidasa (CPGox), en cuyo gen se ha descrito la existencia de 42 mutaciones diferentes (Human Gene Mutation Database, http://www.hgmd.org), mostrando una vez más la naturaleza heterogénea de estas patologías.
Al igual que la PV, es una porfiria mixta en la cual la ocurrencia de ataques agudos es menor que en la PAI. La sintomatología del ataque es similar a los descritos anteriormente. Bioquímicamente se encuentran elevados los valores de ALA, PBG y coproporfirina en orina durante la crisis aguda. También está aumentado el contenido de porfirinas fecales y plasmáticas con un patrón alterado incluso durante la remisión.
PORFIRIAS CUTÁNEAS
El grupo de porfirias cutáneas es heterogéneo, dentro de él se incluyen porfirias hepáticas y eritropoyéticas. Entre las porfirias hepáticas, no agudas, se encuentra la porfiria cutánea tardía (PCT). Entre las porfirias eritropoyéticas se encuentran la protoporfiria eritropoyética (PPE), la porfiria congénita eritropoyética (PCE) y la hepatoeritropoyética (PHE) (Fig 2).
El rasgo común a ellas es una marcada fotosensibilidad cutánea en zonas expuestas a la luz como consecuencia de la presencia de porfirinas circulantes. Entre los signos más frecuentes se encuentran fragilidad cutánea elevada, ampollas, hipertricosis e hiperpigmentación. Todos ellos se intensifican durante la época estival o luego de exposiciones prolongadas a la luz solar.
Bioquímicamente se caracterizan por la acumulación y excreción de porfirinas a través de la orina y/o heces, con un patrón característico para cada porfiria. El contenido de porfirinas plasmáticas está elevado en todos los casos mientras que sólo en las eritropoyéticas se encuentran porfirinas en glóbulos rojos.
Porfiria cutánea tardía:
Esta patología es la más frecuente de todas las porfirias (1:5000 a 1:25000) 2 y se origina como consecuencia de una falla a nivel de la cuarta enzima del metabolismo del hemo, la uroporfirinógeno decarboxilasa (URO-D). Se clasifica en adquirida o esporádica (tipo I) en la cual la actividad de la enzima hepática está disminuida y en familiar o hereditaria (tipo II) 2,19 con actividad enzimática reducida en hígado y eritrocitos. Existe otra forma de PCT familiar en la que sólo la enzima hepática se encuentra alterada (tipo III) 20,21.
La PCT también se ha denominado porfiria turca debido a la aparición de cientos de casos de PCT tipo I en Turquía como consecuencia de una intoxicación masiva con herbicidas policlorados que habían sido empleados en el tratamiento de semillas de trigo usados en la preparación de alimentos.
Como ocurre con otras porfirias se han descrito 70 mutaciones en el gen que codifica para la URO-D (Human Gene Mutation Database, http://www.hgmd.org), indicando nuevamente su heterogeneidad. Se presenta con mayor frecuencia en hombres (4:1) 22.
Las manifestaciones clínicas típicas de la PCT incluyen marcada fragilidad cutánea, hipertricosis malar, hiperpigmentación y lesiones ampollares cuya aparición está frecuentemente asociada a la exposición a factores desencadenantes, entre los cuales encontramos los hidrocarburos aromáticos polihalogenados (hexaclorobenceno, dioxinas), abuso de alcohol, ingestión de estrógenos, sobrecarga de hierro e infecciones con virus de hepatitis C y menos frecuentemente con virus de la hepatitis B 22. Todos estos factores producen disfunción hepática, un signo común en los pacientes con PCT. También se han descrito casos de asociación con portadores del virus de la inmunodeficiencia humana (VIH) 23.
Bioquímicamente se caracteriza por presentar valores elevados de porfirinas alta y parcialmente decarboxiladas, en particular uroporfirina y firiaporfirina, en plasma y orina. Durante la remisión se normaliza tanto el contenido de porfirinas como el patrón de excreción.
Se encuentra descrita una forma recesiva de esta porfiria, en forma homocigota o heterocigota compuesta, denominada PHE. En esta variante familiar extremadamente rara la actividad de Uro-D puede ser entre un 3-27% de su valor normal tanto en eritrocitos como en hígado. Se manifiesta desde la infancia y tiene un fenotipo similar al de la PCE sin eritrodoncia. Hasta el presente sólo se han identificado alrededor de 30 casos.
Protoporfiria eritropoyética:
La PPE es una enfermedad hereditaria producida por fallas en el gen que codifica la ferroquelatasa (FECH) última de las enzimas del camino metabólico del hemo. Su prevalencia en la población general varía entre 1:75000 hasta 1:200000. Se transmite generalmente en forma autosómica dominante aunque existen casos de herencia autosómica recesiva 24,25. En la forma dominante se observaron actividades enzimáticas significativamente menores en pacientes que en portadores, sugiriendo un modo más complejo de herencia. El fenotipo así es el resultado de la combinación de un alelo mutado y un alelo de baja expresión que produce el 50% de la cantidad normal de ácido ribonucleico mensajero (ARNm), confirmado por el estudio de haplotipos en familias PPE 26.
Hasta la fecha se han descripto 90 mutaciones diferentes en el gen que codifica la FECH responsables de la PPE y 7 polimorfismos (Human Gene Mutation Database, http://www.hgmd.org).
Clínicamente se caracteriza por fotosensibilidad temprana, con edema y eritema en áreas expuestas a la luz. Algunos pacientes pueden desarrollar una complicación hepática importante que en un 2% de los casos puede requerir un transplante hepático.
Bioquímicamente se encuentra elevado el contenido de porfirinas, principalmente protoporfirina IX, en glóbulos rojos, plasma y materia fecal. El hígado también acumula protoporfirina IX. Si bien estos parámetros disminuyen con el cuidado y tratamiento, nunca recuperan su valor normal.
Porfiria congénita eritropoyética
La PCE es una rara enfermedad autosómica recesiva de la cual solamente se han descripto alrededor de 200 casos. Se produce por una disminución en la actividad de la Uroporfirinógeno III sintetasa (URO III-S), que cataliza la formación del uroporfirinógeno III, el isómero fisiológico en la biosíntesis del hemo.
Se han identificado 36 mutaciones en el gen que codifica para la UroIII-S (Human Gene Mutation Database, http://www.hgmd.org) y se ha demostrado que la mutación responsable de la patología está relacionada con la severidad del fenotipo.
En la PCE se acumulan grandes cantidades de los isómeros de la serie I, no fisiológico, que se oxidan a uroporfirina I y coproporfirina I provocando la lisis de los glóbulos rojos y la liberación de grandes cantidades de porfirinas que se excretan por orina y materia fecal. La uroporfirina I acumulada en plasma es la principal responsable de las manifestaciones clínicas: lesiones cutáneas severas en zonas expuestas que pueden llevar a la mutilación de los tejidos, hipertricosis, alopecia, eritrodoncia, síntomas que generalmente se presentan desde el nacimiento, aunque se han descrito 13 casos de manifestación tardía 27.
OBJETIVOS
En el presente trabajo se presentan 37 casos de porfirias poco frecuentes que han sido diagnosticados en el Centro de Investigaciones sobre Porfirinas y Porfirias (CIPYP). Para cada uno se describen las características clínicas y bioquímicas que permitieron llegar al diagnóstico preciso, así como también los diferentes tratamientos aplicados y su evolución.
La descripción de estos casos poco frecuentes, sus tratamientos y evolución, facilitarían tanto el diagnóstico diferencial de la porfiria como el conocimiento de las posibilidades terapéuticas en cada caso.
Asimismo para las porfirias heterocigotas con manifestación infantil, su identificación temprana y tratamiento aseguraría una mejor evolución minimizando los riesgos asociados.
CASOS CLÍNICOS
Todos los pacientes estudiados concurrieron o enviaron sus muestras al CIPYP derivados de diferentes Centros de Salud del país con diagnóstico presuntivo de porfiria. Con cada paciente se realizó un interrogatorio acerca del comienzo de la sintomatología aguda y/o cutánea, historia familiar de la enfermedad, exposición a químicos, drogas, infecciones virales (VHC y VHB) y otras patologías y eventuales tratamientos recibidos. De cada paciente se obtuvo consentimiento escrito para su estudio.
MÉTODOS
Para el diagnóstico de la porfiria se realizaron las determinaciones en muestras de orina de 24 horas, materia fecal, glóbulos rojos, sangre y plasma que se indican a continuación, según la metodología descrita por Batlle 28. Las porfirinas plasmáticas (IPP) se determinaron fluorométricamente y las porfirinas en orina (PTO), en eritrocitos (PTS) y en materia fecal (PTMF) se midieron espectrofotométricamente luego de su extracción por cromatografía de intercambio iónico en orina y por solventes orgánicos en los otros dos casos. El patrón de excreción de porfirinas se obtuvo por cromatografía en capa delgada y/o cromatografía líquida de alta presión (HPLC). Los precursores urinarios se cuantificaron por espectrofotometría luego de su extracción por cromatografía de intercambio iónico 28.
La diferenciación entre la PCT tipo I y II se llevó a cabo mediante la determinación de la actividad enzimática de la URO-D eritrocitaria según Afonso 29.
La determinación de la actividad de UROIII-S eritrocitaria se realizó mediante la técnica de ensayo acoplado 30. El análisis del tipo isomérico se realizó por HPLC mediante adaptación de la técnica de Lim 31.
RESULTADOS
PORFIRIAS AGUDAS
Dentro de este grupo de porfirias se presentan 5 casos infanto-juveniles, 2 de PAI, 2 de PV y 1 de CPH.
Porfiria aguda intermitente
La paciente PAI-1 de 12 años de edad es el único caso de PAI infantil diagnosticado hasta el momento en nuestro Centro y en la Argentina. Presentó dolores abdominales, constipación, palpitaciones, astenia, anorexia y orinas oscuras. Previamente había sido medicada con amoxicilina, novalgina y domperidona a raíz de una angina. El análisis de las muestras confirmó el diagnóstico de PAI (Tabla I). Tras graves complicaciones renales la paciente falleció a los pocos días de ser diagnosticada.
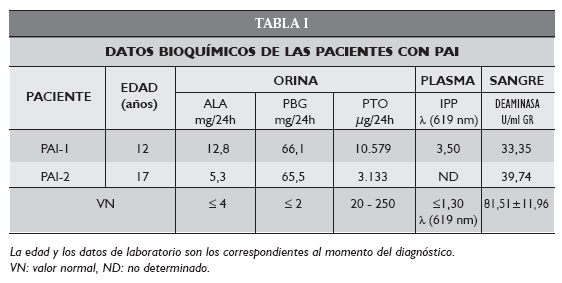
La paciente PAI-2 de 17 años de edad comienza con dolor hipogástrico contínuo, astenia y orinas oscuras. Se realiza un urocultivo, se diagnostica infección urinaria por lo que se le administra gentamina y sertal durante 7 días y fue dada de alta a pesar de la persistencia de los dolores abdominales. A los 18 días presentó cuadriparesia progresiva, dolores musculares generalizados y paresia diafragmática. Fue derivada al hospital María Ferrer donde se le practicó una intubación traqueal y asistencia respiratoria mecánica. A las 12 horas se decidió braquistomizarla. Los resultados bioquímicos confirmaron el diagnóstico de PAI (Tabla I). Como consecuencia de una infección generalizada falleció a los 3 meses del comienzo de la sintomatología.
Porfiria variegata
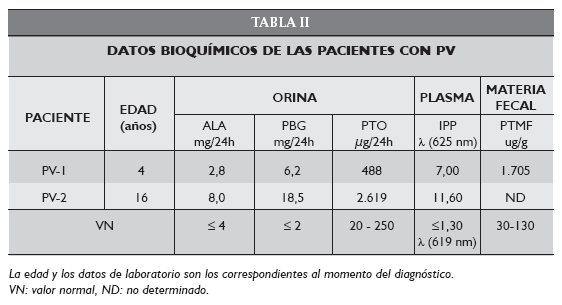
La paciente PV-1 ingresó en el hospital por primera vez a la edad de 4 años, en 1983 con un grave cuadro neurológico y cutáneo. Había sido medicada con fenobarbital por padecer crisis convulsivas febriles recurrentes. Presentaba dolores abdominales, fotosensibilidad, fragilidad cutánea e hipocromías residuales y ampollas en dorso de manos y cara.
La paciente PV-2 de 16 años presentaba síntomas compatibles con un cuadro de porfiria aguda sin sintomatología cutánea. Desde los 13 años manifestaba síntomas de tipo epilépticos por lo cual fue tratada con carbamacepina. El mes anterior a la recepción de las muestras en el CIPYP (febrero de 2006) la paciente había presentado un cuadro de abdomen agudo, por lo que fue laparatomizada, acompañado de debilidad progresiva en miembros superiores e inferiores y algesia. Los electroencefalogramas realizados fueron normales, descartándose el diagnóstico de epilepsia.
Los estudios realizados en orina, sangre, materia fecal y en particular el IPP fueron compatibles en ambos casos con un cuadro de PV (Tabla II).
Coproporfiria hereditaria
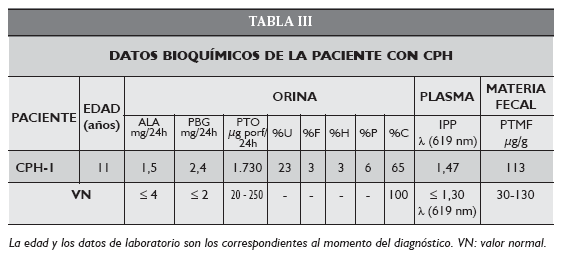
La niña CPH-1 de 11 años residente en la provincia de Jujuy presentaba parálisis cerebral, era espástica y contaba con un historial de infecciones urinarias recurrentes. Debido a sus patologías de base había sido tratada con fenobarbital, valproico, cefalexina, ceftriaxona y gentamicina. Inmediatamente luego del tratamiento con antibióticos había comenzado con orinas oscuras que teñían los pañales y síntomas de dolor neuroabdominal tipo cólico, irritabilidad, sudoración.
Por la sintomatología se llevaron a cabo las determinaciones para una porfiria aguda. Los resultados del análisis de las muestras, enviadas al CIPYP en diciembre de 2005, confirmaron que se trataba de un caso de CPH infantil, el único caso diagnosticado en nuestro Centro y en el país (Tabla III).
PORFIRIAS CUTÁNEAS
Porfiria cutánea tardía
Se presentan los datos de los 25 casos de PCT infantil diagnosticados en nuestro Centro desde 1978 hasta julio de 2006. Los pacientes presentaban sintomatología característica de PCT como ampollas, costras, fragilidad cutánea y fotosensibilidad. El análisis de las muestras confirmó el diagnóstico de PCT (Tabla IV).
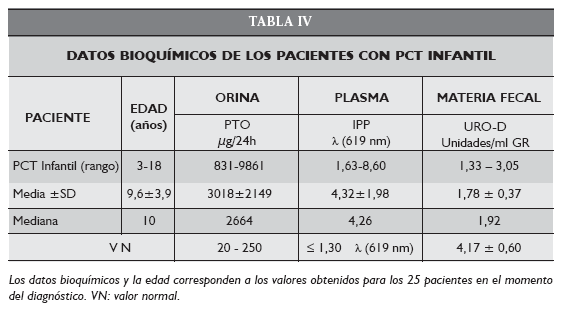
El paciente PHE-1 de 14 años presentaba lesiones ampollares y cicatrizales en piel, en zonas expuestas e hipertricosis desde los 2 años. El caso fue derivado en marzo de 2006 con diagnóstico presuntivo de PCE. Sin embargo los estudios bioquímicos confirmaron, mediante el análisis del patrón de excreción de porfirinas urinarias el diagnóstico diferencial de PHE (Tabla V).
Protoporfiria eritropoyética
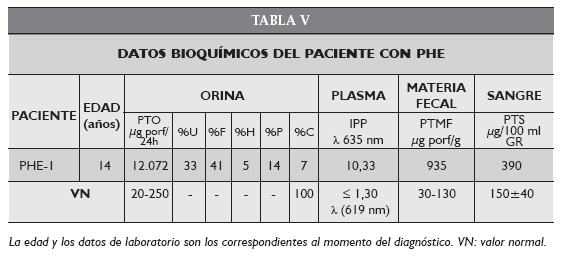
La paciente PPE-1 de 21 años, a pesar de presentar síntomas clínicos de porfiria desde la infancia, no tuvo diagnóstico ni recibió tratamiento. Recién cuando la condición general empeoró, por graves complicaciones hepáticas, vuelve a la consulta en marzo de 2006 y entonces se la confirma como un caso de PPE con una grave falla hepática asociada (hepatitis fulminante autoinmune) (Tabla VI).
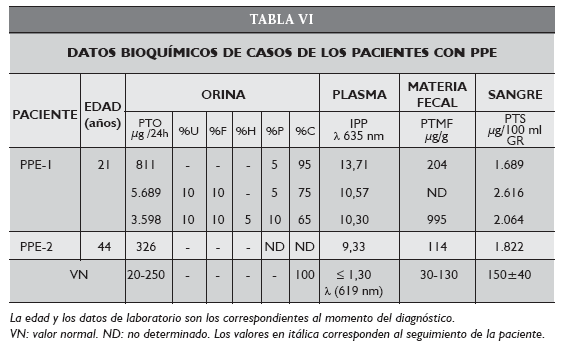
El paciente PPE-2 de 44 años de edad, presentaba un cuadro hepático muy severo en el momento de la derivación de sus muestras al CIPYP (julio de 1999) para confirmar el diagnóstico de PPE (Tabla VI). Si bien la sintomatología cutánea había aparecido desde la primera infancia recién se sospechó una porfiria cuando fue hospitalizado en grave estado.
Porfiria congénita eritropoyética
Los pacientes infantiles PCE-1, PCE-2, PCE-3 y PCE-4 presentaron síntomas de porfiria desde su nacimiento, con excreción muy elevada de porfirinas en orina y materia fecal. El contenido de porfirinas en glóbulos rojos y plasma también fue muy alto (Tabla VII). Los pacientes PCE-1 y PCE-2, eran hermanos y concurrieron por primera vez a la consulta a la edad de 3 y 2 años respectivamente, en el año 1984. La paciente PCE-3 concurrió al CIPYP por primera vez en el año 1994 a los 2 meses de vida. Las muestras del paciente PCE-4 fueron derivadas al CIPYP por primera vez en noviembre de 1998 cuando contaba con sólo 5 meses de edad.
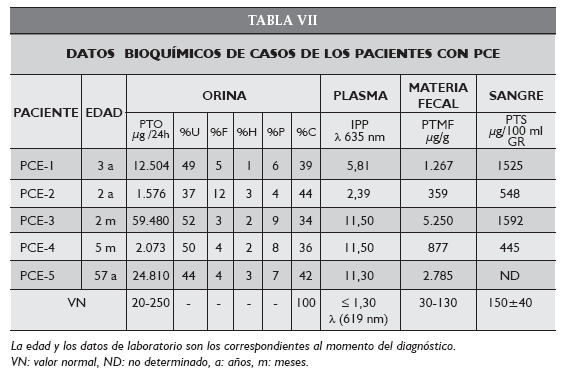
El paciente adulto de 57 años (PCE-5) concurrió por primera vez al CIPYP con diagnóstico presuntivo de PCT en el año 2001 con sintomatología cutánea de 2 años de evolución. A través de los estudios bioquímicos realizados, particularmente el patrón de porfirinas urinarias, se llegó al diagnóstico de PCE (Tabla VII).
El análisis de las porfirinas urinarias por HPLC reveló para PCE-4 y PCE-5 un patrón correspondiente a una PCE (96-98% URO I-2-4% URO III). La actividad de URO III-S en estos casos reveló que la capacidad de la enzima para formar el isómero III estaba disminuida entre un 25-44%.
CONCLUSIONES
La prevalencia de las porfirias varía desde 0,5 a 10 por 100.000 habitantes en diferentes poblaciones 2. En nuestro país la porfiria hepática aguda más frecuente es la PAI con una prevalencia de 1:125.000 seguida de la PV cuya frecuencia es de 1:600.000 32. Dentro de las porfirias cutáneas, la PCT es la porfiria más frecuente en la Argentina (1:37.000) 22 y en segundo lugar se encuentra la PPE con una prevalencia de 1:1.000.000 (resultados no publicados).
Como ya se ha mencionado, las porfirias hereditarias que se manifiestan desde la niñez son la PCE, la PPE y la PHE; sin embargo para la PCE pueden encontrarse casos de manifestación en la edad adulta 25. Por otro lado para la PCT, la PV, la PAI, y la CPH cuya manifestación ocurre generalmente en edad adulta, también se encuentran descritos casos infantiles 2,22. Estos casos corresponden a individuos portadores de la falla genética expuestos a factores desencadenantes exógenos o también a variantes homocigotas, como se ha descrito para la PAI, la PV y la PPE 10,15,16,17,25,33.
En este trabajo se describen 37 casos de porfirias poco frecuentes entre los que se encuentran los casos infantiles de porfirias heterocigotas que incluyen los 25 casos de PCT, un caso de PAI, 2 casos de PV y uno de CPH. En todos ellos los pacientes estuvieron en contacto con drogas porfirinogénicas de diferente etiología que precipitaron el cuadro de porfiria.
Los otros casos corresponden a porfirias homocigotas típicas como los 5 casos de PCE y el de PHE. Se describen además 2 casos severos de PPE.
PORFIRIAS AGUDAS
Porfiria aguda intermitente
Se ha descrito que las complicaciones crónicas de la PAI incluyen enfermedad renal que pude llevar a hipertensión y falla renal 34,35. Una vez diagnosticada la paciente PAI-1 inició el tratamiento con glucosa y vitamina B. A los 4 días presentó un cuadro renal agudo, con mala evolución que desencadenó en un paro cardíaco seguido de muerte.
Porfiria variegata
La paciente PV-1 comenzó con el tratamiento sintomático correspondiente que consistió en administración de suero glucosado al 5% con evidente mejoría clínica y bioquímica a los 4 días de iniciado el tratamiento sin desencadenar nuevas crisis agudas hasta la fecha. Actualmente es un adulto joven y se mantiene asintomática.
El caso de PV-2 es de diagnóstico reciente, cuando se confirma la porfiria y se descarta la epilepsia, comienza el tratamiento para PV con una notable mejoría clínica y bioquímica.
Coproporfiria hereditaria
El análisis de las primeras muestras de la niña confirmaron el diagnóstico de porfiria aguda, suspendiéndose inmediatamente toda la medicación contraindicada y comenzándose el tratamiento con hidratos de carbono indicado para las crisis porfíricas. Los datos bioquímicos obtenidos una vez iniciado el tratamiento mostraron una importante disminución en la excreción de porfirinas urinarias y prácticamente normalización de los valores de los precursores e IPP. Clínicamente la paciente evolucionó favorablemente.
PORFIRIAS CUTÁNEAS
Porfiria cutánea tardía
Si bien la PCT es la más frecuente de las porfirias en toda la población, la PCT tipo II o familiar representa aproximadamente el 25% de los casos totales 22. Es así que la identificación de 25 casos en niños sobre un total de 32 familias con PCT familiar, tanto tipo II ó III, indica un elevado riesgo potencial de manifestación de la porfiria en individuos portadores de corta edad por exposición a factores porfirinogénicos. Entre ellos los hidrocarburos polihalogenados constituyen el principal agente desencadenante de esta porfiria en niños 22.
El tratamiento seguido en estos casos, con muy buenos resultados, es el de la terapia combinada de cloroquina con S-adenosil-Lmetionina junto con una dieta hepatoprotectora 36. Una vez alcanzados los valores normales de porfirinas en orina, se observó remisión de la sintomatología cutánea acompañada de la normalización del patrón de excreción de porfirinas. Los pacientes se mantienen en esta condición si evitan la reexposición a factores desencadenantes.
Dado que no hay bibliografía disponible acerca de la terapia apropiada para aplicar en estos casos el paciente PHE-1, primer caso de esta porfiria en nuestro país, está siendo tratado con pantallas solares y dieta hepatoprotectora con el objeto de minimizar los efectos provocados por la acumulación de porfirinas en los diferentes órganos. Sin embargo, y teniendo en cuenta que una de las posibilidades terapéuticas sería propiciar la eliminación masiva de las porfirinas acumuladas a través de la orina y la materia fecal aplicando terapia con S-adenosil-L-metionina y/o cloroquina como en el caso de otras porfirias, recientemente se ha comenzado a administrarle S-adenosil-L-metionina durante ciclos alternados de 1 mes en una dosis de 20 mg/ kg peso. En este caso particular y ante la presencia de problemas en la visión, se decidió por el momento no administrarle el antimalárico. Los primeros resultados de laboratorio indican un aumento de la eliminación de las porfirinas a través de la materia fecal, que es la vía de excreción que se ve favorecida por la administración de S-adenosil-L-metionina, conjuntamente con una mejoría clínica. El transplante de médula ósea de donante compatible (heterólogo) debe tenerse en cuenta ya que parte del desorden metabólico es eritropoyético.
Protoporfiria eritropoyética
Este desorden hereditario, que se manifiesta desde la niñez, lleva a la acumulación de Proto IX en eritrocitos, plasma, piel e hígado. Alrededor del 35% de los pacientes pueden presentar también alteraciones en la función hepática, mientras que en un 2% de ellos la acción hepatotóxica de la Proto IX causa un grave daño hepático 25. Los dos pacientes que se presentan en este trabajo pertenecen a este grupo.
La paciente PPE-1 fue hospitalizada con un gran deterioro de la función hepática y de su estado general. Además de las lesiones cutáneas, presentaba intensos dolores abdominales y en miembros inferiores. Desde el punto de vista de la porfiria los parámetros bioquímicos analizados estaban todos significativamente alterados, a excepción de ALA y PBG urinarios. Pudo observarse la presencia de una marcada porfirinuria progresiva con alteración del patrón de excreción y del daño hepático (Tabla 7, itálica). Como alternativa terapéutica se decidió someterla a un transplante de hígado, que hasta la fecha no se concretó.
El paciente PPE-2 exhibió sintomatología cutánea desde los 2 años pero su diagnóstico se realizó recién a la edad de 44 años, momento en el cual el daño hepático con cirrosis y colestasis era ya muy serio. Debido a este cuadro se indicó un transplante de hígado que no se llegó a realizar por fallecimiento del paciente.
Porfiria congénita eritropoyética
En la mayoría de los casos esta porfiria se manifiesta desde el nacimiento. Sin embargo se han descrito 13 pacientes adultos 27 con manifestación clínica leve y tardía, a pesar de las elevadísimas cantidades de porfirinas circulantes.
Los pacientes PCE-1 y PCE-2 eran hermanos. Al momento del diagnóstico los valores bioquímicos indicaron una forma clínica más leve para PCE-2. PCE-1 presentó los primeros síntomas a los 2 años, con ampollas en manos, pies y otras áreas expuestas, hipertricosis y eritrodoncia. Dos años más tarde su cuadro empeoró agregando esplenomegalia, razón por la cual fue sometida a una esplenectomía con el objetivo, entre otros, de reducir la intensa hemólisis, lográndose una mejoría transitoria.
La paciente PCE-3 sufría desde el nacimiento de hemorragias generalizadas, por lo que se la sometió a reiteradas transfusiones con sangre entera y plasma, administrándole además vitamina K y antibióticos. A la edad de 1 mes presentaba orinas oscuras y hemorragia fecal y urinaria. El análisis de sangre reveló un hematocrito de 15% con alto porcentaje (1,9%) de reticulocitos concordante con una anemia hemolítica.
Falleció a los 22 meses a consecuencia de la porfiria, evidenciando la severidad de su patología.
El paciente PCE-4, diagnosticado en sus primeros meses de vida, presentó manifestaciones cutáneas leves con ampollas y millia. El tratamiento inicial consistió en la aplicación de pantallas solares. Sin embargo luego se observó un empeoramiento en su condición clínica paralelamente al aumento en la producción y excreción de porfirinas. A los 6 años cuando la manifestación cutánea era más severa fue sometido a un transplante de médula ósea, utilizando como donante compatible su hermana mayor. Los parámetros bioquímicos pre y post transplante, a los 3 y 15 meses de realizado el mismo, mostraron una disminución muy significativa del contenido de porfirinas en todas las muestras analizadas (PTO: 1937-3908 μg/24 h, PTS: 146-265 μg/100 ml GR, PTMF: 124-329 μg/g seco, IPP: 4,10-4,09) en concordancia con una mejoría marcada de su fotosensibilidad y lesiones cutáneas. Para el caso adulto PCE-5, teniendo en cuenta que clínicamente nunca estuvo muy comprometido, se le recomendó como tratamiento el uso de pantallas solares y dieta. El paciente padece también de talasemia menor por lo que recibe ácido fólico.
La determinación de la actividad enzimática en los pacientes PCE-4 y PCE-5 indicó valores de un 60 y 75 % con respecto al normal que fueron superiores a los esperados para una enfermedad autosómica recesiva. Sin embargo, estos resultados se corresponden con la manifestación clínica leve observada en ellos.
El análisis de los casos presentados indica sin lugar a dudas la importancia que un diagnóstico acertado y precoz tiene para el manejo de las porfirias tanto desde el punto de vista del tratamiento apropiado para cada una de ellas, así como para la prevención de la exposición con factores que exacerben la manifestación de la porfiria. Si bien en algunos de los casos más graves la enfermedad es tal que no deja, por el momento, posibilidad de tratamiento, cuando el caso es más leve o es diagnosticado tempranamente existen alternativas terapéuticas de buenos resultados para mejorar la calidad de vida del paciente y cuidar su evolución general.
Es importante también realizar en los miembros de las familias afectadas los estudios genéticos, ya que las determinaciones bioquímicas de rutina permiten llegar a un diagnóstico seguro sólo en los individuos sintomáticos 1. De esta manera la determinación de la mutación en el gen que codifica la enzima deficiente es la forma más precisa de detectar la presencia de casos latentes entre los integrantes de las familias porfíricas.
AGRADECIMIENTOS
El trabajo se llevó a cabo con subsidios del CONICET (2283/89) UBA (UBACYT X 195, X304) y la Agencia de Promoción Científica. Se agradece al Dr. H. Muramatsu y a la Sra. V. Castillo por la atención de los pacientes.
1. Batlle AM del C. Porfirias Humanas. Signos y Tratamientos. En: Porfirias y Porfirinas. Aspectos clínicos, bioquímicos y biología molecular. Actualizaciones Médico Bioquímicas. Ediciones Federación Bioquímica de la Provincia de Buenos Aires. Acta Bioquím Latinoamer 1997; 37-82. [ Links ]
2. Anderson KE, Sassa S, Bishop DF y Desnick RJ. Disorders of heme biosynthesis. X-linked sideroblastic anemia and the porphyrias. En: Scriver CR, Beaudet AL, Sly WS, Valle D Editores. The Metabolic and Molecular Bases of Inherited Diseases, 8th Edición. New York: McGraw-Hill 2000; 2961-3062. [ Links ]
3. Elder GH. Hepatic Porphyrias in children. J Inherit Metab Dis 1997; 20: 237-246. [ Links ]
4. Kauppinen R. Porphyrias. Lancet 2005; 365: 241-252. [ Links ]
5. Barclay N. Acute intermittent porphyria in childhood: a negleted diagnosis? Arch Dis Child 1974; 49: 404-405. [ Links ]
6. Beauvais P, Klein ML y Denave L. Porphyria ague intermittente a l'age de quatre mois. Aped 1976; 33: 987-992. [ Links ]
7. Picat C, Delfau M, De Rooij FW, Beukeveld GJ, Wolthers BG, Wadman SK, Nordmann Y y Grandchamp B. Identification of the mutation in the parents of a patient with a putative compound heterozygosity for acute intermittent porphyria. J Inherit Metab Dis 1990; 13: 684-686. [ Links ]
8. Llewellyn DH, Smith SJ, Elder GH, Hutchesson AC, Rattenbury JM y Smith MF. Homozygous acute intermittent porphyria: compound heterozygosity for adjacent base transitions in the same codon of the porphobilinogenase gene. Hum Genet 1992; 89: 97-98. [ Links ]
9. Xu W, Solís C, Bermejo AM y Desnick RJ. Clinical and molecular delineation of severe homozygous dominant acute intermittent porphyria. Pediatr Res 1995; A155: 916. [ Links ]
10. Solis C, Martínez-Bermejo A, Naidich TP, Kaufmann WE, Astrin KH, Bishop DF y Desnick RJ. Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias. Arch Neurol 2004; 61:1764-1770. [ Links ]
11. Hessels J, Voortman G, Van der Wagen A, Van der Elzen C, Scheffer H y Zudderhoudt J. Homozygous acute intermittent porphyria in a 7-year-old boy with massive excretions of porphyrins and porphyrin precursors. J Inherit Metab Dis 2004; 27: 19-27. [ Links ]
12. Hift RJ, Meissner PN, Todd G, Kirby P, Bilsland D, Collins P, Ferguson y Moore MR. Homozygous variegate porphyria: an evolving clinical syndrome. Postgrad Med J 1993; 69: 781-786. [ Links ]
13. Frank J, McGrath J, Lam H, Graham RM, Hawk JLM y Christiano AM. Homozygous variegate porphyria: identification of mutations on both alleles of the protoporphyrinogen oxidase gene in severely affected probands. J Invest Dermatol 1998; 110: 452-455. [ Links ]
14. Roberts AG, Puy H y Dailey TA. Molecular characterization of homozygous variegate porphyria. Hum Mol Genet 1998; 7: 1921-1925. [ Links ]
15. Corrigall AV, Hift RJ, Davids LM, Hancock V, Meissner D, Kirsch RE y Meissner PN. Homozygous Variegate Porphyria in South Africa: Genotypic Analysis in Two Cases. Mol Genet Metab 2000; 69: 323-330. [ Links ]
16. Kauppinen R, Timonen K, von und zu Fraunberg M, Laitinene E, Ahola H, Tenhunen R, Taketani S y Mustajoki P. Homozygous variegate Porphyria: 20 y follow-up and characterization of molecular defect. J Invest Dermatol 2001; 116: 610-613. [ Links ]
17. Palmer RA, Elder GH, Barrett DF y Keohane SG. Homozygous variegate Porphyria: a compound heterozygote with novel mutations in the protoporphyrinogen oxidase gene. Br J Dermatol 2001; 144: 866-869. [ Links ]
18. Poblete-Gutiérrez P, Wolff C, Farías R y Frank JA. Chilean boy with severe photosensitivity and finger shortening: the first case of homozygous variegate porphyria in South America. Br J Dermatol 2006; 154: 368-371. [ Links ]
19. De Vernuil H, Atiken G y Nordmann Y. Familial and sporadic porphyria cutanea tarda: two different diseases. Hum Genet 1978; 44: 135-151. [ Links ]
20. Elder GH, Sheppard DM, Enriquez de Salamanca R y Olmos A. Identification of two types of porphyria cuatnea tarda by measurement of erythrocyte uroporphyrinogen de carboxylase: an exception to the rule?. Dermatologica 1989; 178: 206-208. [ Links ]
21. D'Alessandro GL, Grisso D, Macri A, Biolcati G y Topi GC. Familial porphyria cutanea tarda with normal erythrocytic urodecarboxylase: an exception to the rule? Dermatologica 1989; 178: 206-208. [ Links ]
22. Méndez M, Rossetti MV, Batlle AM del C y Parera VE. The role of inherited and acquired factors in the development of porphyria cutanea tarda in the Argentinean population. J Am Acad Dermatol 2005; 52: 417-424. [ Links ]
23. Melito VA, Parera, VE, Rossetti MV y Batlle A. Manifestación de porfiria cutánea tardía en pacientes infectados con el virus de la inmunodeficiencia humana. Acta Bioquím Clin Latinoam 2006; 40: 29-34. [ Links ]
24. Sarkany R y Cox T. Autosomal recessive erythropoietic protoporphyria: a syndrome of severe photosensitivity and liver failure. Q J Med 1995; 88: 541-549. [ Links ]
25. Whatley SD, Mason NG, Khan M, Zamiri M, Badminton MN, Missaoui WN, Dailey TA, Dailey HA, Douglas WS, Wainwright NJ y Elder GH. Autosomal recessive erythropoietic protoporphyria in the United Kingdom: prevalence and relationship to liver disease. J Med Genet 2004; 41: 105. [ Links ]
26. Gouya L, Martin-Schmitt C, Robreau AM, Austerlitz F, Da Silva V, Brun P, Simonin S, Lyoumi S, Grandchamp B, Beaumont C, Puy H y Deybach JC. Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. Am J Hum Genet 2006; 78: 2-14. [ Links ]
27. Kontos AP, Ozog D, Bichakjian C y Lim HW. Congenital erythropoietic porphyria associated with myelodisplasia presenting in a 72-year-old: report of a case and review of the literature. Br J Dermatol 2003; 148: 160-164. [ Links ]
28. Batlle AM del C. El Laboratorio de las Porfirias, en Porfirias y Porfirinas. Aspectos clínicos, bioquímicos y biología molecular. Actualizaciones Médico Bioquímicas. Ediciones Federación Bioquímica de la Provincia de Buenos Aires. Acta Bioquím Latinoamer 1997; 145 - 171. [ Links ]
29. Afonso SG, Chinarro S, Stella AM, Lenczner JM, Batlle AM del C y Magnin PH. Uroporfirinógeno decarboxilasa eritrocitaria y hepática en Porfiria Cutánea Tardía. Rev Argent Dermatol 1985; 66: 12 -23. [ Links ]
30. Tsai SF, Bishop DF y Desnick RJ. Purification and properties of uroporphyrinogen III synthase from human erythrocytes. J Biol Chem 1987; 262: 1268- 1273. [ Links ]
31. Lim C K, Rideout J M y Wright D J. Highperformance liquid chromatography of naturally occurring 8-, 7-, 6-, 5- and 4- carboxylic porphyrin isomers. J Chromatogr 1983; 282: 629-641. [ Links ]
32. Parera VE, De Siervi A, Varela LS, Rossetti MV y Batlle A. Acute Porphyrias in the Argentinean population. A review. Cell Mol Biol 2003; 49: 493-500. [ Links ]
33. Poh-Fitzpatrick MB, Wang X, Anderson KE, Bloomer JR, Bolwell B y Lichin AE. Erythropoietic protoporphyria: altered phenotype after bone narrow transplantation for myelogeneous leukemia in a patient heteroallelic for ferrochelatase gene mutations. J Am Acad Dermatol 2002; 46: 861-866. [ Links ]
34. Andersson C y Lithner F. Hypertension and renal disease in patients with acute intermittent porphyria. J Intern Med 1994; 236: 169-175. [ Links ]
35. Andersson C, Wikbergh B, Stegmayr B y Lithner F. Renal symptomatology in patients with acute intermittent porphyria. A population-based study. J Int Med 2000; 248: 319-325. [ Links ]
36. Batlle AM del C, Stella AM, Kaminsky AR, Kaminsky C y Guzmán H. Two cases of infantil Porphyria Cutanea Tarda. Succesful treatment with oral SAdenosyl-L-Methionine and low dose of oral chloroquine. Br J Dermatol 1987; 116: 407-415. [ Links ]

