










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista argentina de dermatología
versão On-line ISSN 1851-300X
Rev. argent. dermatol. v.91 n.1 Ciudad Autónoma de Buenos Aires jan./mar. 2010
ARTÍCULOS ORIGINALES
Lesiones dermatológicas del síndrome de Apert. A propósito de un caso clínico
Dermatological Features Associated With Apert Syndrome. Apropos Of A Case
* Profesor en Ciencias Médicas. Profesor Asociado Cátedra de Farmacología. Universidad Católica de Cuyo. Villa Mercedes. San Luis. Dermatólogo Universitario. Universidad de Buenos Aires.
** Profesor en Ciencias Médicas. Profesor Titular Cátedra de Salud Pública. Universidad Católica de Cuyo. Villa Mercedes. San Luis. Médico Cirujano Pediátrico. Universidad Nacional de Cuyo.
Córdoba 372. Villa Mercedes. San Luis.
e-mail: canechi01@hotmail.com
Presentamos una paciente de 26 años de edad con Síndrome de Apert (SA) quien presenta lesiones acneiformes desde hace trece años, caracterizadas por la rebeldía al tratamiento y una extraordinaria tendencia a la recidiva.
Como este cuadro es realmente raro, consideramos oportuno realizar una revisión bibliográfica de la patología dermatológica asociada al mismo.
PALABRAS CLAVE: Síndrome de Apert; Dermatosis asociadas al Síndrome de Apert.
SUMMARY
We report a woman 26 years old with Apert Syndrome (AS). She has acneiform lesions from thirteen years ago, characterized by resistence to treatment and a remarkable tendency to recur.
As this picture is really weird, we consider it timely to review literature about dermatologic disease associated with it.
KEY WORDS: Apert´s Syndrome; Dermatological features in Apert Syndrome.
INTRODUCCIÓN
Fueron Baumgartner en 1842 (Krankenphysionomie, Stuttgart) y luego Wheaton en 1894 (Trans Path Soc London), los primeros en referirse a esta anomalía, pero es Apert 1 quien primero describió la tríada integrada por hipoplasia maxilar, craneosinostosis y sindactilia. 2, 3
Aunque este síndrome puede ser heredado en forma dominante, la mayoría de los casos son esporádicos y en éstos, la edad avanzada de los padres jugaría algún rol. 4, 5
La rareza de esta patología está demostrada por los datos epidemiológicos disponibles, su incidencia varía entre 1: 50.000 nacimientos 6, y 1: 160.000 7 según las series. En regiones de EE.UU la prevalencia del Síndrome de Apert (SA) es de 12.4 por millón de nacimientos. 8 Algunos autores consideran que de todos los pacientes con craneosinostosis, el 4,5% corresponde al SA.
CASO CLÍNICO
Paciente de 26 años de edad que desde los 13 presenta lesiones, diagnosticadas como acné polimorfo en tórax, cara y espalda. Fue tratada con tetraciclinas (minociclina) 100 mg diarios, durante períodos variables obteniéndose alguna mejoría. Recientemente se le indicó tretinoína 20 mg por día durante 90 días, pero la familia suspendió esta terapia por razones de índole económico y la escasa mejoría observada.
OBJETIVO
Realizar una meticulosa revisión bibliográfica de este síndrome, sin embargo, cumplir este objetivo fue dificultoso, pues la mayoría de las publicaciones aluden a casos únicos, siendo entonces difícil obtener conclusiones generales.
COMENTARIOS
Las mutaciones responsables del SA se localizan en el gen ubicado en 10q26, gen conocido como "Factor Receptor 2 del Crecimiento Fibroblástico" (FR2CF) 9, 10, 11, sin embargo no es el único síndrome relacionado con mutaciones de este gen, ya que, se sabe que otras alteraciones del mismo dan lugar a los síndromes de Crouzon, Pfieffer y Saethe-Chotzen.
El trastorno comienza a expresarse en los primeros meses de la lactancia (antes de los tres meses de vida), aunque se ha sugerido que la inhibición del crecimiento en la zona esfeno-frontal y en la sutura coronaria comienza muy precozmente en la vida fetal; esto ocasiona finalmente el cierre prematuro de la sutura coronaria (Gráfico I), ocasionando una gran falla anatómica observable en la línea media del cráneo la cual se extiende desde la zona glabelar hasta la fontanela posterior, incluidas la sutura metópica, la fontanela anterior y la sutura sagital. La anómala expresión del gen FR2CF provoca que un mayor número de células precursoras entren en osteogénesis, esto origina un aumento de la matriz ósea subperióstica y a posteriori osificación prematura del cráneo. El orden y el porcentaje de la sinostosis en estas suturas determinan el grado de deformidad y discapacidad. Una vez cerrada la sutura, el crecimiento del cerebro perpendicular a la misma se limita, pero el desarrollo cerebral continúa a expensas del resto de las suturas abiertas, aunque las complejas y múltiples sinostosis, causan hipoplasia medio-facial, órbitas hundidas, alteraciones del dorso nasal, hipoplasia maxilar (Fig 1) y a veces obstrucción de las vías respiratorias superiores. 12 La forma definitiva del cráneo en esos pacientes es variable, puede ser acrocefalia, braquicefalia, turribraquicefalia, frente elevada y prominente, occipucio plano, casi un 4% de los lactantes con SA presenta una rara anomalía denominada cráneo en trébol. En 1966 Schauerte y St Aubin señalaron que la sinostosis en el SA es progresiva, proponiendo denominarlo: "sinostosis progresiva y sindactilia". Blank en el año 1960 en una revisión distinguió dos categorías clínicas: la acrocefalosindactilia "típica" (Síndrome de Apert), cuyo signo distintivo es una mano medio digital en masa, constituida por la fusión de los dígitos 2 al 4 (Fig 2), con una sola uña compartida y la acrocefalosindactilia "atípica", donde se agrupan los pacientes sin esta malformacion.13

Gráfico I: las líneas rojas indican la orientación de la craneosinostosis.

Fig 1: frente abombada, proptosis.

Fig 2: pulgar malformado, sindactilia de los dígitos 2 al 4. La separación parcial entre anulares y medios es quirúrgica.
Las alteraciones de la región de la boca, nariz, garganta, oído y ojos incluyen una boca en reposo de forma trapezoidal (Fig 3), implantación baja de las orejas, ocasionalmente pérdida congénita de la audición conductiva. Fisuras palpebrales inclinadas, hipertelorismo, órbitas hundidas, proptosis, exoftalmos, estrabismo, ambliopía, atrofia óptica, queratocono, glaucoma congénito y ocasionalmente papiledema. La nariz tiene un puente nasal notablemente deprimido, corto y ancho con punta bulbosa con apariencia de pico de loro y puede presentar estenosis o atresia de las coanas, prognatismo, arcos palatinos elevados, úvula bífida y paladar fisurado (Fig 4).

Fig 3: hipoplasia maxilar, nariz en "pico de loro". Boca de forma trapezoidal.

Fig 4: hallux corto, y sindactilia total (ósea y cutánea) del segundo al quinto dedo de ambos pies.
Los problemas de ortodoncia incluyen superposición de los dientes superiores, mala oclusión, retraso en la dentición, erupción dentaria ectópica, incisivos en forma de pala y dientes supernumerarios (Fig 5). 8,14
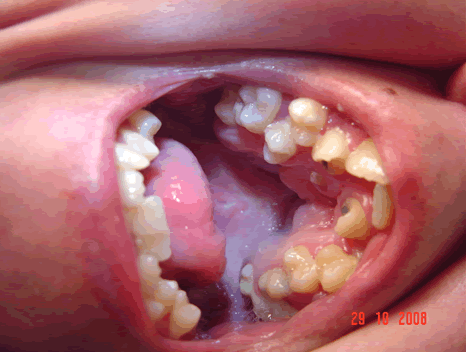
Fig 5: paladar fisurado reparado quirúrgicamente. Mala implantación dentaria.
Las anomalías del SNC más comunes incluyen megalocefalia, agenesia del cuerpo calloso, estructuras límbicas malformadas, ventrículomegalia de grado variable y encefalocele. El nivel de inteligencia varía desde la normalidad hasta la deficiencia mental. En un estudio realizado se encontró que el 48% de los pacientes tenían un coeficiente mental entre normal y fronterizo, el 44% mostraba un retraso mental variable desde leve a moderado y el 2% exhibía un retraso mental severo. 15 Separados entre sí por 70 años, otros autores, vieron que la mayoría de los pacientes con este síndrome eran mentalmente retrasados. 16, 17. Pelz y col y Reiner y col (citados por Papp), notaron que el 39,3% de los niños con SA residentes con sus familias exhibían un coeficiente intelectual normal; en contraste, esta buena condición solo fue observada en el 12,5% de los niños alojados en instituciones especializadas. 18
Casi el 68% de los pacientes con SA, presentan sinartrosis de las vértebras cervicales, frecuentemente C5 y C6, signo útil para distinguir a estos pacientes de aquellos afectados por la enfermedad de Crouzon, donde las sinartrosis, además de ser más raras (solo el 25% la tienen), suelen estar localizadas en otro sitio (C2 y C3). 19
ALTERACIONES DERMATOLÓGICAS
1- Hiperhidrosis 20
Es el resultado de la disfunción de las glándulas apócrifas. En un estudio de 136 pacientes se la pudo constatar en el 100% de los casos 21; este aumento de la sudoración suele ser más ostensible en axilas, con una característica que la hace más desagradable, el mal olor (bromhidrosis). Los intentos infructuosos para atenuarla han incluido en el pasado hasta técnicas quirúrgicas. 22
2- Lesiones acneiformes
Solomon las describió primero en el año 1971 23; otros han notado la persistencia de sus brotes, algunos muy severos y resistentes a los tratamientos habituales. Recientemente se obtuvieron buenas respuestas con isotretinoína y por separado con anticonceptivos orales. 24
La historia natural de esta anomalía comienza en la adolescencia o tiempo después, cuando la piel comienza a tornarse oleosa, para luego aparecer pápulas foliculares similares al acné, pero con una diferencia respecto del "clásico": las pápulas no solo se ubican en cara y tronco sino también en antebrazos, glúteos y muslos, incluso se las ha observado adoptando una distribución "nevoide".
Ciertas mutaciones somáticas del Factor de Crecimiento de los Queratinocitos (RFCQ) determina un mosaicismo epidérmico, el cual provocaría anomalías del aparato pilosebáceo, o un aumento de la sensibilidad a los andrógenos de la unidad folículo-sebácea, o ambos fenómenos combinados, cuyo producto final es el brote acneiforme. 25,26, 27, 28, 29.30, 31, 32, 33, 34, 35
3- Sinoniquias
Esta alteración suele afectar más a las manos que a los pies y la asociación de falanges distales con sinoniquia, puede ser hallada en las manos pero nunca en los pies. 36
4- Uñas quebradizas
La causa de esta anomalía en el SA no ha sido establecida todavía, se sabe además que existen otras enfermedades, congénitas o no, que la exhiben. Dentro de las congénitas se cuentan el Síndrome de Hutchinson Gilford y la displasia ectodérmica con ectrodactilia.
5- Interrupción del trazado normal de las cejas
Está motivado por un defecto óseo subyacente, el cierre precoz de la sutura esfenoparietal, lo que crea una lámina orbitaria del frontal muy corta; esto provoca a su vez una gran retracción y elevación supraorbitaría, más pronunciada lateralmente, resultando en una solución de continuidad del trazado normal de las cejas. 37,38
6- Hipopigmentación de la piel, pelo y ojos (albinismo óculo-cutáneo)
Casi el 27% de los pacientes con SA presentan hipopigmentación de la piel, pelo y ojos. 39,40,41,42 De nueve pacientes con SA observados por Margolis y col cinco mostraban esta anomalía 43. El color del pelo variaba desde castaño claro a rubio, la piel era pálida y los írides eran verdosos o azulados, habiéndose demostrado que la causa de esta hipopigmentación es la falta de migración de los melanoblastos in útero. Se ha observado además transiluminación del iris e hipopigmentación del fundus, asociado con ausencia o disminución de los reflejos foveales. No se demostró nistagmo pendular ni disminución de la agudeza visual, diferente a lo que sucede en la mayoría de las formas del albinismo óculo-cutáneo. Las pruebas indican que la pérdida de la pigmentación y las anomalías esqueléticas en el SA son el resultado de alteraciones independientes, genéticamente relacionadas, de un proceso sucedido en un punto común, durante la gestación.
7- Hiperqueratosis plantar lateral
Esta es debida a la fusión ósea progresiva de los huesos del tarso y metatarso, lo que ocasiona transferencia del peso corporal hacia la región media y lateral del pie; el acto de caminar y estar de pie provoca luego el aumento de espesor de la piel. 44
8- Perionixis
Se observa con mayor frecuencia en pacientes institucionalizados y suele localizarse más habitualmente en las uñas de los pies que en las manos. La colonización periungueal por cándidas estaría favorecida por la existencia de sinoniquia, uñas quebradizas e hiperhidrosis.
9- Arrugas excesivas en la piel que cubre la frente
Consecuencia de la hipoplasia ósea del hueso frontal, la frente en el SA suele ser abultada e inclinada; dicha arquitectura y la ley de la gravedad, serian las responsables del exagerado arrugamiento de la región frontal (Cohen y col, 1995).
10- Hoyuelos (depresiones o fositas) en los nudillos, hombros y codos
Pueden aparecer desde la infancia, siendo esta localización poco frecuente; su localización es más típica en cara o salientes óseas, aunque se han observado fositas escapulares supraespinosas congénitas aisladas o asociadas al síndrome de la Trisomía 9p, o el Síndrome Russell-Silver, o en la delección del cromosoma 18q 45 o en el Síndrome del pterigión poplíteo y en el Síndrome de los hoyuelos autosómicos dominantes 46,47, 48, 49. Es necesario destacar que en algunas infecciones tales como la rubéola congénita, 50,51,52, en algunos traumas mecánicos y más raramente en formas de hipofosfatemias adquirida, se pueden observar estas fositas. 53, 54
En Argentina se ha observado un paciente con una placa alopécica en cuero cabelludo y pápulo-pústulas en antebrazos y dorso; la histología de estas lesiones mostró foliculitis y perifoliculitis granulomatosa. 55
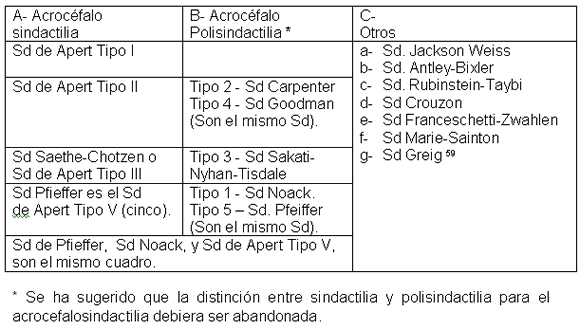
Los diagnósticos diferenciales han sido resumidos en el Cuadro I, debe aclararse sin embargo que muchas de estas entidades, aparentemente, distintas, son consideradas ahora variantes alélicas. 56, 57, 58,59
CUADRO I
Diagnósticos diferenciales del Síndrome de Apert
RESULTADOS
De han descrito diez signos dermatológicos en el SA, de los cuales dos, la hiperhidrosis y el acné son constantes. En un solo caso autores argentinos refieren la aparición de una placa alopécica en cuero cabelludo, pápulo-pústulas en antebrazos y dorso, cuyo significado se desconoce.
CONCLUSIONES
Como las manifestaciones del SA pueden ser lentas, no siempre es diagnosticado temprano y si bien es probable que otras especialidades médicas sean consultadas antes que Dermatología, es de buena práctica que el dermatólogo esté informado de las anomalías asociadas al Síndrome de Apert.
1. Apert E. De l'Acrocephalosyndactylie. Bull Soc Med 1906; 23: 1310. [ Links ]
2. Gorlin RJ, Pindborg JJ, Cohen Jr MM. Syndromes of the Head and Neck. Editado por Mc Graw-Hill. Segunda Edición. New York. USA. 1976; 32-35. [ Links ]
3. Sedano H, Sauk J, Gorlin RJ. Oral Manifestations of Inherited Disorders. Editado por Butterworth Group. Primera Edición. Boston. USA. 1977; 88-89. [ Links ]
4. Gorlin RJ. Branchial arch and oro-acral disorders. In: Gorlin JJ, Cohen MM Jr y Levin LS Editores. Syndromes of the Head and Neck. Oxford Univ Press 1990; 3: 641–649. [ Links ]
5. Allanson JE. Germinal mosaicism in Apert Syndrome. Clin Genet 1986; 29: 429-433. [ Links ]
6. Rollnick BR. Male transmission of Apert syndrome. Clin Genet 1988; 33: 87-90. [ Links ]
7. Albuquerque MA, Cavalcanti MG. Computed tomography assessment of Apert syndrome. Pesqui Odontol Bras 2004; 18 (1): 35-39. [ Links ]
8. Blank CE. Apert´s Syndrome (a type of acrocephalosyndactyly): Observations on a British series of thirty nine cases. Ann Hum Genet 1960; 24: 151-164. [ Links ]
9. Tolarova M, Harris J A, Ordway D E, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents' age, and ethnicity in Apert syndrome. Am J Med Genet 1997; 72: 394-398. [ Links ]
10. Mc Kusick V A. Apert Syndrome. www.ncbi.nlm.nih.gov. 1986. [ Links ]
11. Wilkie AOM, Slaney S F, Oldridge M, Poole M D, Ashworth G J, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, Malcolm S, Winter RM, Reardon W. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nature Genet 1995; 9: 165-172. [ Links ]
12. Stanley SF, Oldridge M, Hurst JA, Morriss-Kay GM, Hall CM, Poole MD, Wilkie AOM. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet 1996; 58: 923-932. [ Links ]
13. Chen H. Apert Syndrome. www.emedicine.com. 2007. [ Links ]
14. Maroteaux P, Fonfria MC. Apparent Apert syndrome with polydactyly: rare pleiotropic manifestation or new syndrome?. Am J Med Genet 1987; 28: 153-158. [ Links ]
15. Schauerte EW, St-Aubin PM. Progressive synosteosis in Apert's syndrome (acrocephalosyndactyly): with a description of roentgenographic changes in the feet. Am J Roentgen 1966; 97: 67-73. [ Links ]
16. Patton MA, Goodship J, Hayward R, Lansdown R. Intellectual development in Apert's syndrome: a long term follow up of 29 patients. J Med Genet 1988; 25: 164-167. [ Links ]
17. Park EA, Powers G F. Acrocephaly and scaphocephaly with symmetrically distributed malformations of the extremities. Am J Dis Child 1920; 20: 235-315. [ Links ]
18. Cohen MM Jr, Kreiborg S. The central nervous system in the Apert syndrome. Am J Med Genet. 1990; 35: 36-45. [ Links ]
19. Papp H E. Síndrome de Apert (Acrocefalosindactilia). Presentación de dos Casos Clínicos. Acta Odontol Venez 1999; 37 (3): 163-167. [ Links ]
20. Kreiborg A, Barr M Jr, Cohen M M Jr. Cervical spine in the Apert syndrome. Am J Med Genet 1992; 43: 704-708. [ Links ]
21. Cohn MS y Mahon MJ. Apert's syndrome (acrocephalosyndactyly) in a patient with hiperhidrosis. Cutis 1993; 52 (4): 205-208. [ Links ]
22. Cohen MM Jr, Kreiborg S. Visceral anomalies in the Apert syndrome. Am J Med Genet 1993; 45: 758-760. [ Links ]
23. Hess J, Lonergan I, Rozzelle AA, Arneja JS. Axillary Osmidrosis in Apert Syndrome: Management With an Arthroscopic Shaver Technique. J Craniofacial Surg 2008; 19 (4): 1126-1130. [ Links ]
24. Solomon LM, Cohen MM Jr, Pruzansky S. Pilosebaceus abnormalities in Apert-type acrocephalosyndactyly. Birth Defects Orig Artic Series 1971; 7: 193- 195. [ Links ]
25. Hsieh T, Ho N. Resolution of acne following therapy with an oral contraceptive in a patient with Apert syndrome. J Am Dermatol 2005; 53 (I): 173-174. [ Links ]
26. Cuerda E, del Pozo J, Rodríguez-Lozano J, Peña-Penabad C, Fonseca E. Acne in Apert's syndrome: treatment with isotretinoin. J Dermatol Treat 2003; 14 (1): 43-45. [ Links ]
27. Steffen C. The acneiform eruption of Apert's syndrome is not acne vulgaris. Am J Dermatopathol. 1984;6 (3):213-220. [ Links ]
28. Henderson CA, Knaggs H, Clark A, Highet AS, Cunliffe WJ. Apert's syndrome and androgen receptor staining of the basal cells of sebaceous glands. Br J Dermatol 1995; 132 (1): 139-143. [ Links ]
29. Hivnor MCM, Yan AC, Honig PJ. Acne Arising in an Epidermal Nevus. Pediatr Dermatol 2007; 24 (5): 534-535. [ Links ]
30. Verma SMD, Draznin M. Apert´s syndrome. Dermatol Online J 2005; 11 (1): 15. [ Links ]
31. Downs AM, Condon CA, Tan R. Isotretinoin therapy for antibiotic-refractory acne in Apert's syndrome. Clin Exp Dermatol 1999; 24 (6): 461-463. [ Links ]
32. Mc Naughton PA, Rodman OG. Apert síndrome. Cutis 1980; 25: 538-540. [ Links ]
33. Steffen C. Acneiform eruption in Apert syndrome. Acrocephosyndactyly. Arch Dermatol 1982; 118: 206-208. [ Links ]
34. Munro CS, Wilkie AO. Epìdermal mosaicism producing localized acne: somatic mutation in FGFR2. Lancet 1998; 352: 704-705. [ Links ]
35. Campanati A, Marconi B, Penna L, col. Pronounced and early acne in Apert syndrome: a case successfully treated with oral isotretinoin. Eur J Dermatol 2002; 12: 496-498. [ Links ]
36. Cuerda E, Del Pozo J, Rodríguez-Lozano J, col. Acne in Apert syndrome. J Dermatol Treat 2003; 14: 43-45. [ Links ]
37. Apert syndrome. www.apert-syndrome.de. 2005. [ Links ]
38. Cohen MM Jr, Kreiborg S. Visceral anomalies in the Apert syndrome. Am J Med Genet 1993; 45: 758-760. [ Links ]
39. Cohen MM Jr, Kreiborg S. Hands and feet in the Apert syndrome. Am J Med Genet 1995; 57: 82-96. [ Links ]
40. Boissy RE, Hoath S. Congenital Patterned Leukodermas. http://www.emedicine.com. 2007. [ Links ]
41. Freiman A, Tessler O y Barankin B. Apert syndrome. Int J Dermatol 2006; 45 (11): 1341-1343. [ Links ]
42. Cohen MM Jr, Kreiborg S. Cutaneous manifestations of Apert syndrome. Am J Med Genet 1995; 31: 58-94. [ Links ]
43. Awasthy N, Gupta H. Apert Syndrome. Pediatr Oncall [serial online] 2005; 1: 2. [ Links ]
44. Margolis S, Siegel IM, Choy A, Breinin GM. Oculocutaneous albinism associated with Apert's syndrome. Am J Ophthalmol 1977; 84 (6): 830-839. [ Links ]
45. Narang T, Kanwar AJ, Dogra S. Brachycephaly and syndactyly: Apert's syndrome. Indian J Dermatol Venereol Leprol [serial online] 2008; 74: 395-396. [ Links ]
46. Ghidoni PD, Hale DE, Cody JD, Gay CT, Thompson NM, McClure EB, col. Growth hormone deficiency associated in the 18q deletion syndrome. Am J Med Genet 1997; 3: 7-12. [ Links ]
47. http://www.thedoctorsdoctor.com/diseases/skin_dimples.htm [ Links ]
48. Sáez-de-Ocariz M, Durán-McKinster C, Orozco-Covarrubias L, Ruíz- Maldonado R. Hoyuelos supraespinosos escapulares congénitos: a propósito de dos casos pediátricos. Dermatol Pediatr Lat 2005; 3 (2): 149-151. [ Links ]
49. Virgili A, Tosti G, Bettoli V, Corazza M. Multiple congenital symmetric skin dimples. Dermatology 2002; 204: 293-295. [ Links ]
50. Spencer JM, Schneiderman PI, Grossman ME. Bilateral skin dimples on the shoulders. Pediatr Dermatol 1993; 10 (1): 16-18. [ Links ]
51. Cohen MM Jr, Kreiborg S. Skeletal abnormalities in the Apert syndrome. Am J Med Genet 1993; 47: 624-632. [ Links ]
52. Hall BD. Skin dimples and rubella. Br J Clin Pract. 1968; 22: 193-194. [ Links ]
53. Hammond K. Skin dimples and rubella. Pediatrics 1967; 39: 291-292. [ Links ]
54. Wood VE. Congenital fossae about the shoulder. Plast Reconstr Surg 1990; 85: 798-800. [ Links ]
55. Wendling D, Blanc D, Hory B. Skin dimples in hypophosphatasia. Dermatologica 1989; 178: 179-180. [ Links ]
56. Cohen Sabban E, Gruber M, Soljancic C, Coronell S, Ruiz Begurie J, Casas J, Larralde M. Síndrome de Apert, manifestaciones cutáneas. Presentación de un caso. Dermatol Argent 2003; 3: 168-173. [ Links ]
57. Goodman RM, Sternberg M, Shem-Tov Y, Katznelson MB, Hertz M, Rotem. Acrocephalopolysyndactyly type IV: a new genetic syndrome in 3 sibs. Clin Genet 1979; 15 (3): 209–214. [ Links ]
58. Cohen DM, Green JG, Miller J, Gorlin RJ, Reed JA. Acrocephalopolysyndactyly type II-Carpenter syndrome: clinical spectrum and an attempt at unification with Goodman and Summit syndromes. Am J Med Genet 1987; 28 (2): 311–324. [ Links ]
59. Ernyei S. Craniofacial dysplasia associated with congenital cataract, impairment of hearing and brachydactyly. Am J Ophthalmol 1966; 62 (4): 697- 702. [ Links ]

