










Servicios Personalizados
Revista

Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de dermatología
versión On-line ISSN 1851-300X
Rev. argent. dermatol. vol.92 no.1 Ciudad Autónoma de Buenos Aires ene./mar. 2011
REVISIÓN
Púrpura de Schönlein Henoch – Qué hay de nuevo?
Henoch Schönlein purpura - What about new?
MC Mazas *
* Carrerista en Dermatología Pediátrica. Universidad de Buenos Aires.
Hospital General de Niños "Ricardo Gutiérrez". Gallo 1330 (1425) Ciudad Autónoma de Buenos Aires.
e-mail: ceciliamazas@speedy.com.ar
RESUMEN
La púrpura de Schönlein Henoch es la vasculitis más frecuente en la infancia, aunque puede verse en adultos. Se caracteriza por la presencia de púrpura palpable, dolor abdominal o articular que puede acompañarse de hematuria. El compromiso renal es más común en adultos. Su diagnóstico es clínico aunque la biopsia puede ser útil para descartar otros diagnósticos diferenciales.
Se comentan en esta revisión los cambios realizados en cuanto a los criterios diagnósticos y las novedades en relación a la fisiopatología.
PALABRAS CLAVE: Vasculitis; Vasculitis leucocitoclástica; Púrpura de Schönlein Henoch; Hematuria.
SUMMARY
Henoch Schönlein purpura is the most common vasculitis in childhood, althoug it can be seen in adults. It is caharacterized by the presence of palpable purpura, abdominal or joint pain may be accompanied by hematuria. Renal involvement is more common in adults. The diagnosis is clinical but biopsy may be useful to rule out other differential diagnoses.
Discussed in this review the changes made regarding the diagnostic criteria and developments in relation to pathophysiology.
KEY WORDS: Vasculitis; Leukocytoclastic vasculitis; Henoch Schönlein purpura; Hematuria.
INTRODUCCIÓN
La Púrpura de Schönlein Henoch (PSH) es una de las vasculitis más comunes en la infancia, afecta frecuentemente al grupo etario comprendido entre los dos y seis años, aunque puede presentarse en niños mayores e incluso en adultos.
En 1802 William Heberden publicó el caso de un niño de cinco años con edema, artralgias, hematuria, dolor abdominal acompañado de melena y pápulas en sus miembros inferiores. Años más tarde, en 1837, Johann Schönlein describe la asociación de artralgia y púrpura. Su discípulo Eduard Henoch fue quien reconoció el compromiso intestinal y posteriormente el renal como parte de este síndrome. A partir de ahí toma el nombre de Púrpura de Schönlein-Henoch 1.
EPIDEMIOLOGÍA E INCIDENCIA
La PSH presenta una incidencia en chicos de 10.22 /100.000 por año. Más del 90% de los casos reportados son pacientes pediátricos menores de diez años, con un pico de incidencia a los seis años de edad 2-6.
La PSH se presenta como un cuadro leve en los lactantes y niños menores de dos años y de forma más grave en adultos, con riesgo mayor de causar enfermedad renal a largo plazo 2. En relación a este punto hay un artículo de la revista Pediatric Neonatology: "Clinical Manifestations and Outcomes of Henoch-Schönlein Purpura: Comparison between Adults and Children" de Shih-Pin Hung y col, pertenecientes al Department of Pediatrics, National Taiwan University Hospital, Taipei, Taiwan y al Department of Pediatrics, Cathay General Hospital, Taipei, Taiwan. Este estudio retrospectivo de cinco años tuvo como objetivo comparar las diferencias, en relación a la epidemiología, parámetros clínicos y datos de laboratorio, entre niños y adultos con PSH en Taiwan. También, analizó los factores asociados al mal pronóstico de la nefritis por PSH. En dicho trabajo se expresa una incidencia en adultos de 1.3/100.000, con una media de cincuenta años. Todos los pacientes en ambos grupos presentaron púrpura palpable, que involucraba el tronco y las extremidades superiores en aproximadamente el 50% de los pacientes. La hematuria macroscópica y el edema de miembros inferiores fueron más frecuentes en el adulto. Alrededor de un tercio de los niños, pero ninguno de los adultos, sufrieron sinusitis. Los pacientes del grupo pediátrico presentaron con mayor frecuencia dolor abdominal, antes que apareciera la erupción púrpura. El intervalo entre la aparición del dolor abdominal y la erupción cutánea purpúrica, podría ser tan extenso como veintiún días (rango 1-21 días, media 8,31 ± 5,52 días). Todos los pacientes en el grupo infantil desarrollaron hematuria a las dos semanas (33% en la primera semana y 67% en la segunda), mientras que este intervalo puede ser de hasta cinco semanas en los adultos (34% dentro de dos semanas y el 67% durante la quinta semana). La frecuencia de hematuria grave y edema de extremidades inferiores, también fue significativamente mayor en pacientes adultos que en niños.
En cuanto a la comparación de los resultados de laboratorio observaron una frecuencia elevada de glóbulos blancos (recuento de leucocitos >11.000/mm3), significativamente mayor en el grupo pediátrico. La cantidad de pacientes que presentaron compromiso fue de catorce (21,54%) en el grupo pediátrico. Doce de ellos presentaron hematuria aislada y/o proteinuria, mientras que sólo dos pacientes un síndrome nefrótico. Sin embargo, todos los pacientes de este grupo tuvieron función renal normal. En cambio, doce pacientes del grupo adulto (52,6%) presentaron afectación renal, seis de ellos con insuficiencia (tres presentaron IR sumada a proteinuria y hematuria y los otros tres IR junto con síndrome nefrótico). Los pacientes con dolor abdominal en el inicio de la enfermedad, tuvieron una probabilidad mayor de desarrollar el síndrome nefrótico.
Este estudio, por los análisis estadísticos de OR e IC95%, reveló que la edad >20 años, género masculino, heces con sangre, erupción cutánea persistente (definida como la erupción de purpúricas que persiste más de un mes) y las recaídas de los síntomas eran pobres indicadores pronósticos para la nefritis HSP. Todos los pacientes con PSH presentaron erupción purpúrica como síntoma más común de inicio, tanto en adultos como en niños. Sin embargo, el dolor abdominal se presentó como el síntoma previo a la púrpura más común en los niños. Ninguno de los pacientes tenía nefritis antes de la aparición de la púrpura. La frecuencia de afectación gastrointestinal fue más común en los niños de este estudio, pero García-Porrúa y col informaron que las manifestaciones gastrointestinales fueron similares en ambos grupos etarios.
Este estudio también encontró que en los adultos con nefritis HSP, frecuentemente se presenta con síntomas después del mes de iniciada la actividad HSP, mientras que en los pacientes pediátricos sucede dentro de las dos semanas de comenzado el cuadro. A pesar de la presencia de orinas normales, se sugiere el seguimiento de por lo menos seis meses principalmente en los adultos, por la predisposición a padecer complicaciones renales. El riesgo de progresión de la enfermedad a la insuficiencia renal osciló entre 5-15% en los niños y fue de aproximadamente el 30% en adultos 8.
El riesgo de insuficiencia renal es doce veces mayor, si la presentación inicial se complica por el síndrome nefrítico o nefrótico, en lugar de sólo una orina patológica. Además, el 19% de los adultos presentó graves manifestaciones renales, el 13% desarrolló insuficiencia renal y el 10% de ellos requirió hemodiálisis. En oposición, el paciente pediátrico generalmente presenta una recuperación completa.
Los pacientes con dolor abdominal como síntoma inicial de la enfermedad, tenían una probabilidad significativamente mayor de desarrollar el síndrome nefrótico y la erupción cutánea persistente es un indicador de mal pronóstico para la nefritis HSP 7.
FISIOPATOLOGÍA
En los pacientes con PSH, los complejos de inmunoglobulina A (IgA) se depositan en los pequeños vasos generando la clínica característica, petequias y púrpura palpable. Cuando estos complejos autoinmunes se depositan en los pequeños vasos de la pared intestinal, desencadenan hemorragias que pueden ir de leve a severa (Fig 1). Si esto mismo ocurre en los vasos del mesangio renal puede producir glomérulonefritis.
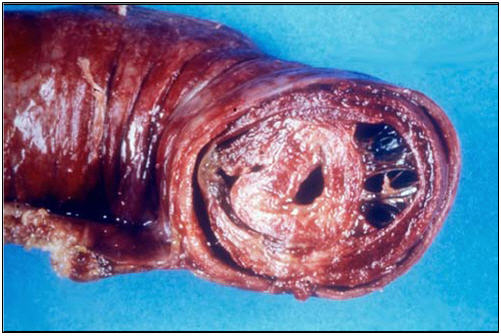
Fig1: sección del intestino grueso que demuestra la invaginación intestinal. Tomada de Gastroenterol Clin North Am 1998; 27 (4): 747-782.
La exposición a un antígeno, generalmente secundario a una infección, medicamentos u otros factores ambientales puede desencadenar la formación de anticuerpos e inmunocomplejos. De los gérmenes, el estreptococo del grupo A es el que frecuentemente ha sido encontrado en más del 30% de los niños con nefritis secundaria a PSH; los títulos de anticuerpos antiestreptolisina O tienen más probabilidades de ser positivos en pacientes con PSH y nefritis 6.
Recientemente, se encontró un receptor de plasmina asociado a la nefritis (NAPlr) al que se le uniría un antígeno de estreptococo, ubicado en el mesangio glomerular de los niños con nefritis por PSH 12. Estos datos sugieren la participación del agente infeccioso en la iniciación y/o evolución del cuadro.
Otros agentes infecciosos desecadenantes son: Parvovirus B19, Bartonella henselae, Helicobacter pylori, Parainfluenza, Coxsackie, Adenovirus, la Hepatitis A y B, Micoplasmas, Virus de Epstein-Barr, Varicela, Campylobacter.
Los complejos de IgA se forman y se depositan en la piel, intestino y glomérulos, provocando una respuesta inflamatoria localizada. La vasculitis leucocitoclástica se desarrollará posteriormente, con la necrosis de los vasos de calibre pequeño. Normalmente la IgA se encuentra en suero y secreciones mucosas, tiene dos isotipos: IgA1 y IgA2. En la mucosa la IgA es IgA2 con un 60% en forma polimérica, mientras que en suero es IgA1 y el 90% monomérica.
En la PSH los complejos se forman con IgA1 poliméricos. Una forma anormal de la IgA1 conocida como Gal-d IgA1 (refiriéndose a una deficiencia de galactosa de la O-enlazados glicanos en la región bisagra de IgA1) ha sido identificada altamente en la nefritis por HSP nefritis en comparación con HSP sin nefritis 11.
En cuanto al mecanismo fisiopatológico, Yao-Hsu Yang y col del National Taiwan University Hospital 13, proponen cuatro hipótesis que pueden ser desencadenadas por infecciones.
En primer lugar, la hipótesis del mimetismo molecular, es decir, los microbios pueden compartir epítopos con los de los pequeños vasos sanguíneos de los humanos. Al invadir estos patógenos el cuerpo humano se desencadenaría, a raíz de una reacción cruzada, una respuesta inflamatoria humoral y celular en los pequeños vasos. En segundo lugar, la hipótesis de activación "bystander", donde los patógenos al generar inflamación inespecífica y daño celular, pondrían al descubierto antígenos que habitualmente no están expuestos al sistema inmunológico. Tercero, se postula la hipótesis de autoalteración, donde los agentes infecciosos interactúan con las proteínas de los vasos, generando nuevos antígenos que activarían la reacción inflamatoria. Por último, la cuarta hipótesis es la que postula la presencia de superantígenos, donde algunas bacterias y virus se transforman en superantígenos, sin la necesidad de procesamiento y presentación por células presentadoras, interactuando directamente con las células T.
Es probable que no haya un único patógeno que pueda desarrollar una PSH y que muchos microbios pueden desencadenar una vía común para inducir la inflamación de los pequeños vasos. Yao-Hsu Yang y col del National Taiwan University Hospital 13 concluyen en presentar el siguiente modelo:
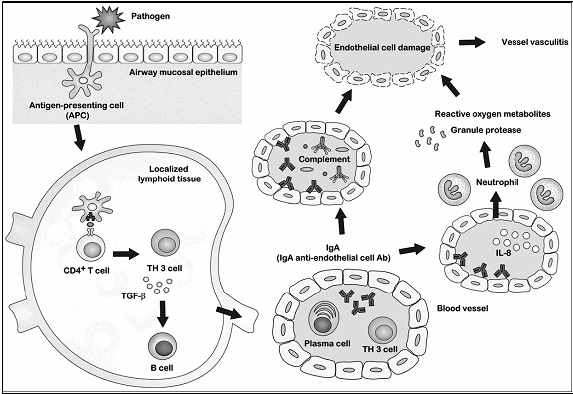
Gráfico I: Modelo hipotético para el desarrollo de la PSH
Para aquellos individuos con un perfil genético específico, la introducción de microbios a través de la mucosa de las vías respiratorias, puede activar un subgrupo de células T que a través de las células presentadoras de antígenos, tomarían los microorganismos y/o sus antígenos y migrarían al tejido linfoide localizado en el que presentan epítopos de células T CD4 +. Desde allí un subconjunto de TGF-β que secretan las células T CD4 + estimulan la proliferación de las células TH3, las que posteriormente activan las células B. Las células B activadas reconocen el mismo antígeno e inducen la diferenciación a células productoras de IgA, aumentando los niveles de IgA sérica. Algunos anticuerpos circulantes IgA presentan una reacción cruzada con las células endoteliales (IgA AECA) y dañan las células mediante la activación de la vía alterna del complemento; también pueden estimular directamente las células endoteliales para producir IL-8, que recluta y activa los neutrófilos para causar aún más daño en las células endoteliales, a través de metabolitos directos como el oxígeno reactivo y proteasas de gránulos.
El objetivo de este trabajo fue proporcionar una perspectiva adicional sobre el mosaico de la autoinmunidad.
CLÍNICA
La aparición de púrpura palpable, dolor abdominal y artritis pueden presentarse en cualquier orden, aunque el dolor abdominal y la artritis no están universalmente presentes. La progresión de los síntomas puede darse en el transcurso de días o en forma insidiosa en semanas 14. Estos síntomas pueden acompañarse de fatiga y fiebre baja.
Todos los pacientes con PSH desarrollan una erupción, que comienza con pápulas eritematosas no pruriginosas o urticaria que evoluciona a púrpura palpable.
La púrpura se define como hemorragias cutáneas mayores a 10 mm de diámetro, que no desaparecen a la vitropresión. El cambio de color en las lesiones antes de desaparecer, puede llevar un período de aproximadamente diez días. La erupción se localiza generalmente en las zonas de declive o de mayor presión, tales como las extremidades inferiores, las nalgas y en la superficie extensora de las extremidades.
La artritis no migratoria ocurre en el 75 % de los pacientes con PSH, comprometiendo rodillas, tobillos y más comúnmente las pequeñas articulaciones. Los síntomas de la artritis incluyen inflamación, calor y limitación en la movilidad. Estos síntomas son transitorios, no dejan deformidad y puede preceder a la púrpura en un 15 a 25 % 4,5.
El dolor abdominal ocurre en 60-65% de los pacientes, puede simular un dolor abdominal agudo confundiéndose con cuadro de apendicitis 15. El dolor es típicamente cólico, se produce alrededor de una semana después de la aparición de la erupción. Los vómitos y la hemorragia gastrointestinal (oculta y manifiesta) se desarrollan en un 30% 16. Es poco común que la hemorragia se presente en forma severa, pero de serlo requerirá estudios de mayor complejidad para su valoración.
También puede presentarse con un hematoma mural con riesgo de invaginación 16.
La enfermedad renal es la secuela más grave, ocurre en un 40 a 50% 17. Aunque la muerte por PSH es rara, la enfermedad renal es la principal causa de muerte en estos pacientes. El riesgo de padecer compromiso renal es superior en: mayores de diez años, aquellos con púrpura persistente, dolor abdominal severo o episodios recidivantes 18.
A diferencia del dolor abdominal o de la artritis, que puede preceder a la erupción, la enfermedad renal es una secuela tardía. Por lo general comienza dentro del primer mes y rara vez se produce más de seis meses después de la enfermedad 15. Los signos de compromiso renal son: hematuria microscópica y proteinuria. La enfermedad renal suele remitir de forma espontánea en la mayoría de los pacientes. Sin embargo, la glomérulonefritis progresiva puede desarrollarse en los pacientes con proteinuria persistente.
A modo de resumen se presenta el siguiente Cuadro 19:


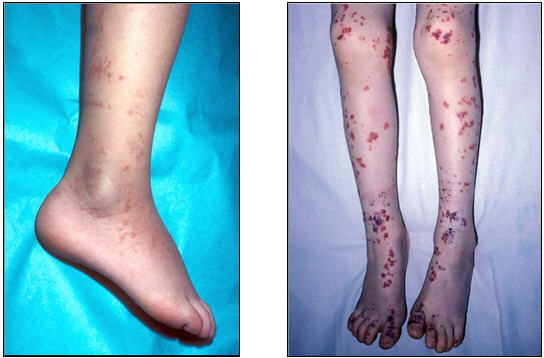
Figs 2 y 3: ejemplos de distribución de púrpura palpable en niños.
DIAGNÓSTICO
No existe ninguna prueba definitiva para realizar el diagnóstico de PSH, el mismo es clínico y debe sospecharse en aquellos pacientes que presentan la tríada clásica: púrpura palpable no trombocitopénica, dolor abdominal y artritis.
La biopsia es útil para descartar otros diagnósticos. En la PSH es útil para mostrar la característica vasculitis leucocitoclástica y los depósitos de IgA.
La biopsia renal mostrará una glomérulonefritis membranoproliferativa similar a la nefropatía por IgA 10. En 1990 11, el Colegio Americano de Reumatología definió los criterios definidos para el diagnóstico de PSH.
Para realizar el diagnóstico se requería la presencia de dos de las cuatro características. Tuvo una sensibilidad de diagnóstico de 87,1% y una especificidad del 87,7%.
Dichos criterios fueron: pacientes de 20 años o menores, púrpura palpable sin trombocitopenia, compromiso intestinal de tipo difuso o diagnóstico de isquemia intestinal, cambios histológicos que mostraran granulocitos en las paredes de las pequeñas arteriolas y vénulas (vasculitis leucocitoclástica).
En 2005, se publicó The International Consensus Conferencerealizado en Viena, donde se adaptaron los criterios diagnósticos a la población pediátrica. Los mismos quedaron definidos por: púrpura palpable como característica obligatoria, acompañado de dolor abdominal, artritis, compromiso renal y biopsia que muestre los depósitos de IgA. Se eliminó el criterio de edad. Estos criterios han sido aceptados por las organizaciones de expertos, pero aún se espera la validación en los ensayos clínicos. (Ver el siguiente Cuadro):

En 2009, se publicó en la revista Clinical Rheumatology 21 un estudio longitudinal de la microvascularización, estudiada por capilaroscopía en chicos con PSH, con el objetivo de describir por la participación de la microvasculatura y los cambios capilares en los niños con PSH, intentando establecer una relación con la clínica. Para ello evaluaron treinta y un pacientes que padecían PSH con videomicroscopía durante la fase aguda y después de seis meses comparados con un grupo control. Se observaron anormalidades morfológicas así como la desorganización en un alto porcentaje. El edema fue una constante y las macrohemorragias sólo se encontraron en tres niños.
Las características capilaroscópicas (cambios en la arquitectura y morfología) en fase aguda, fueron estadísticamente significativas en comparación con los controles sanos, aunque no fue posible revelar un patrón específico para PSH. La normalización de las anomalías vistas en la capilaroscopía fueron resueltas después de seis meses, pero todavía persistía el edema en más de la mitad de los pacientes. Este dato podría apoyar la observación anterior de Martino y col 22, que confirma que las alteraciones observadas en la microangiopática durante la fase aguda, no presentan resolución a pesar de la remisión clínica. No obstante, la presencia de un edema leve en los niños sanos, podría sugerir que este es uno de los signos de los capilares en proceso de maduración. La evaluación del edema es todavía controvertida y hasta el momento no se ha logrado un acuerdo en el análisis de este parámetro.
La capilaroscopía puede ser una herramienta simple para evaluar anomalías microvasculares en la fase aguda de PSH y la persistencia de edema podría sugerir una enfermedad en resolución incompleta, demostrado a nivel microvascular.
LABORATORIO
Los estudios de laboratorio complementan la clínica y permiten evaluar riesgo de complicaciones y seguimiento.
Los estudios básicos incluyen: hemograma completo con recuento de plaquetas, pruebas de función renal, análisis de orina, coagulograma y dosaje de los niveles de IgA 5,12,15, acompañados de exudado de fauces o dosaje de antiestreptolisina-O, sangre oculta en materia fecal y biopsia de piel.
Los estudios de imagen no se realizan de forma rutinaria en la evaluación de la PSH.
Cuando el compromiso intestinal es severo, tal vez pueda ser necesario recurrir a la arteriografía o la endoscopía para evaluación del cuadro.
Ante los cuadros de dolor abdominal importante puede requerirse una ecografía abdominal o una tomografía computarizada y si se piensa en un cuadro de invaginación intestinal, puede utilizarse el enema de bario 23.
DIAGNÓSTICOS DIFERENCIALES
Dentro de los diagnósticos diferenciales podemos encontrar 2:
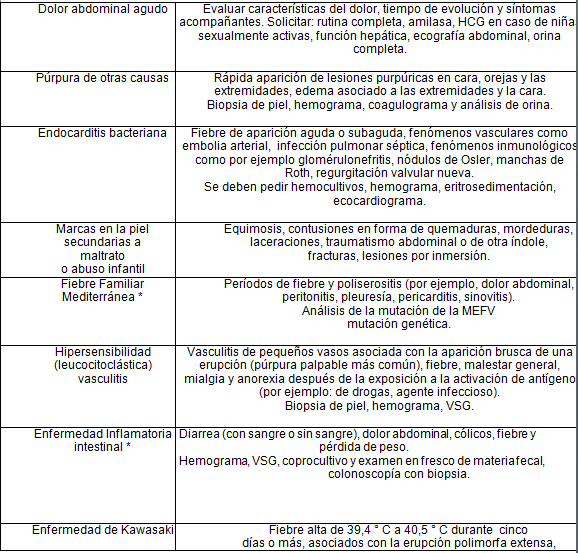
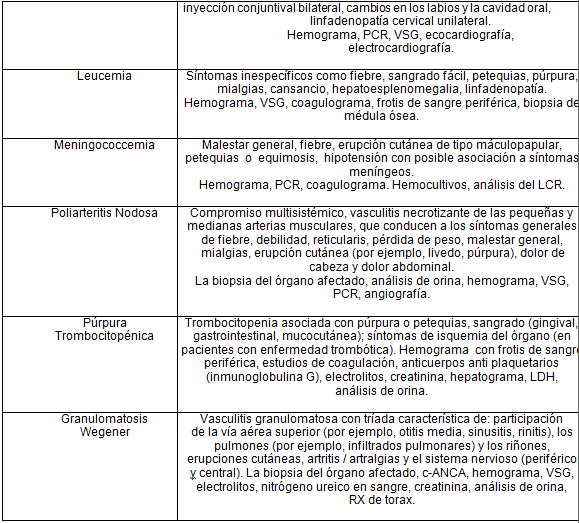
* Puede coexistir con la PSH.
TRATAMIENTO
Dado que la PSH resuelve espontáneamente en el 94% de los niños, sólo se realiza tratamiento de sostén. El mismo consiste en reposo y analgesia en caso de dolor abdominal o artralgias 10. Los AINEs pueden agravar los síntomas gastrointestinales y deben evitarse en pacientes con conocida afectación renal.
El reposo relativo y la elevación de las extremidades afectadas durante la fase aguda de la enfermedad, pueden ayudar a prevenir la púrpura. Los pacientes deben ser informados sobre la posibilidad, que pueden presentar púrpura recurrente al aumentar su nivel de actividad. La hospitalización puede estar indicada, cuando no se cuenta con un adecuado control ambulatorio, ante la presencia de edema y dolor escrotal o de cuero cabelludo, deshidratación, hemorragia, dolor abdominal o cuando hay compromiso renal importante con insuficiencia significativa.
El tratamiento temprano con esteroides es el más apropiado para los niños con afectación renal o síntomas graves. La administración de prednisona oral de 1 a 2 mg por kg al día durante dos semanas, se ha utilizado para tratar el dolor abdominal de moderado a grave, el compromiso articular importante y para acelerar la resolución de la PSH en niños.
Un ensayo doble ciego aleatorizado halló que el tratamiento precoz con prednisona, reduce el dolor abdominal y articular en los niños. Aunque la prednisona no previene la enfermedad renal, fue útil en el tratamiento de la misma después que esta comenzó 24.
En meta-análisis publicado en Pediatrics 2007 25, se encontró que los corticosteroides utilizados en niños con PSH, redujeron la media de tiempo de resolución del dolor abdominal y disminuyeron las probabilidades de desarrollar enfermedad renal persistente. Esta terapéutica no afecta a la resolución de la púrpura 25.
El tratamiento temprano y agresivo se recomienda para niños y adultos con insuficiencia renal grave 24,25,26. Las opciones de tratamiento incluyen esteroides a dosis altas con inmunosupresores, altas dosis de inmunoglobulina intravenosa, plasmaféresis y trasplante renal.
Un estudio reciente encontró que la ciclofosfamida fue eficaz en los pacientes con nefropatía manifiesta 26.
SEGUIMIENTO
La mayoría de los pacientes se recuperan completamente dentro de las cuatro semanas. Las recidivas pueden ocurrir hasta en un tercio de los pacientes, dentro de los primeros seis meses después de la aparición y son más comunes en pacientes con insuficiencia renal 15.
El pronóstico a largo plazo depende de la gravedad de la afectación renal.
Una revisión sistemática reveló que la aparición de la enfermedad renal en pacientes con PSH, se desarrolló en el plazo de cuatro semanas en el 85% de los pacientes, de seis semanas en el 91% y seis meses en el 97% 27. La insuficiencia renal permanente no se desarrolló en pacientes con orina normal, aunque ocurrió en el 19,5% de los pacientes con síndrome nefrítico o nefrótico 27. La medición de la presión y el seguimiento con orinas, debe realizarse al momento del diagnóstico y durante el seguimiento, con el objetivo de pesquisar el compromiso renal. Si el análisis de orina inicial es normal o si hay hematuria aislada sin síndrome nefrítico o nefrótico, debe realizarse un análisis de orina mensual durante los primeros seis meses después del diagnóstico 27.
A modo de resumen se presenta un algoritmo publicado en The European Journal of Pediatrics 2009 1:
Seguimiento de todos los pacientes con HSP (para identificar aquellos con compromiso renal).
(Chicos con cuadro de recurrencia deben ser monitoreados como si fuera su primer episodio).
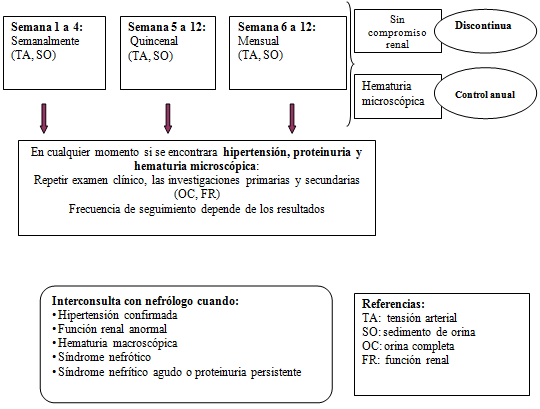
PSH Y EMBARAZO
El embarazo puede verse comprometido en aquellas mujeres que tuvieron PSH en la infancia 1. Un estudio de seguimiento a largo plazo de niñas que sufrieron PSH en la infancia, mostró que dieciseis de cuarenta y cuatro embarazos de término fueron complicados, secundariamente a la aparición de proteinuria e hipertensión 28. Estos hallazgos fueron repetidos en otro estudio a largo plazo, en el que el seguimiento mostró que dieciseis de veintitres embarazos tuvieron complicaciones 29.
Es recomendable que las mujeres que padecieron PSH durante la infancia realicen un seguimiento cercano de su embarazo, principalmente en lo que se refiere a la pesquisa de hipertensión arterial y búsqueda de proteinuria.
CONCLUSIONES
-
Tener presente que la PSH es la vasculitis más frecuente en Pediatría, con una incidencia anual de 10.22/100.000 por año, no sucediendo lo mismo en la población adulta que muestra una incidencia de 1.3/100.000.
-
La exposición a un antígeno, generalmente secundario a una infección, medicamentos u otros factores ambientales puede desencadenar la formación de anticuerpos e inmunocomplejos. De los gérmenes, el estreptococo del grupo A es el más frecuente aunque se han demostrado otros.
-
Es probable que no haya un único patógeno que pueda desarrollar una PSH y que muchos microbios pueden desencadenar una vía común, para inducir la inflamación de los pequeños vasos.
-
El diagnóstico es clínico.
-
No existe ninguna prueba definitiva para realizar el diagnóstico de PSH.
-
Todos los pacientes con PSH presentan púrpura palpable. Este es un criterio clínico indispensable para el diagnóstico, como bien se definió en el Consenso de Viena en el 2005.
-
La aparición de púrpura palpable, dolor abdominal y la artritis puede ser en cualquier secuencia.
- García - Porrúa y col mostraron que la frecuencia de graves manifestaciones renales en niños, fueron significativamente menores que en los adultos y que una recuperación completa ocurrió en la mayoría de los casos. La enfermedad renal es una secuela tardía, lo que marca la importancia del seguimiento a largo plazo de la función renal, para detectar precozmente el compromiso de dicho órgano. Se propone que este seguimiento debiera realizarse durante un año, en aquellos que no hayan presentado hematuria y toda la vida en quienes se hubiera presentado algún tipo de compromiso renal.
-
En el estudio de la revista Pediatric Neonatology "Clinical Manifestations and Outcomes of Henoch-Schönlein Púrpura: Comparison between Adults and Children", por los análisis estadísticos de OR e IC95%, se marcó que la edad >20 años, género masculino, heces con sangre, erupción cutánea persistente (definida como la erupción de purpúricas que persiste más de un mes) y las recaídas de los síntomas, eran pobres indicadores pronósticos para la nefritis PSH.
- La utilización de la capilaroscopía como método no invasivo, para sumar datos clínicos de diagnóstico. A través de la misma pueden observarse cambios en la arquitectura y morfología en fase aguda. Aunque todavía no es posible revelar un patrón específico para PSH.
-
Dado que la PSH resuelve espontáneamente en el 94% de los niños, sólo se realiza tratamiento de sostén. El mismo consiste en reposo y analgesia en caso de dolor abdominal o artralgias.
-
Para el dolor abdominal importante se sugiere la utilización de corticoides junto con protección gástrica.
-
Tratamiento agresivo en aquellos con compromiso renal. Un meta-análisis publicado en Pediatrics 2007 25, encontró que los corticosteroides usados en niños con PSH redujeron la media de tiempo de resolución de dolor abdominal y disminuyeron las probabilidades de desarrollar enfermedad renal persistente. Esta terapéutica no afecta a la resolución de la púrpura 25.
- Es recomendable que las mujeres que tuvieron PSH durante la infancia, realicen un seguimiento cercano de su embarazo, principalmente en lo que se refiere a pesquisa de hipertensión arterial y búqueda de proteinuria.
1. Mc Carthy HJ y col. Diagnosis and Managment of Henoch-Schönlein purpura. Eur J Pediatr 2009; 169: 643-650. [ Links ]
2. Reamy BV y col. Henoch-Schönlein Purpura. http://www.aafp.org/afp. [ Links ]
3. Gardner-Medwin JM y col. Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002; 360 (9341): 1197-1202. [ Links ]
4. Calviño MC y col. Henoch-Schönlein purpura in children from northwestern Spain: a 20-years epidemiologic and clinical study. Medicine 2001; 80 (5): 279-290. [ Links ]
5. Trapani S, Micheli A, Grisolia F y col. Henoch-Schönlein purpura in childhood: epidemiological and clinical analysis of 150 cases over a 5-year period and a review of the literature. Semin Arthritis Rheum 2005; 35 (3): 143-153. [ Links ]
6. Saulsbury FT. Epidemiology of Henoch Schönlein purpura. Cleve Clin J Med 2002; 69 (2): S II87-SII90. [ Links ]
7. Shih-Pin Hung y col. Pediatric Neonatology, Clinical Manifestations and Outcomes of Henoch-Schönlein Purpura: Comparison between Adults and Children. Correspondiente al: Department of Pediatrics. National Taiwan University Hospital. Taipei - Taiwan y al: Department of Pediatrics. Cathay General Hospital. Taipei - Taiwan. [ Links ]
8. Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol 2001; 13 (1): 35-40. [ Links ]
9. Allen AC, Willis FR, Beattie TJ y Feehally J. Abnormal IgA glycosylation in Henoch-Schönlein purpura restricted to patients with clinical nephritis. Nephrol Dial Transplant 1998; 13 (4): 930–934. [ Links ]
10. Gedalia A. Henoch-Schönlein purpura. Curr Rheumatol Rep 2004; 6 (3):195-202. [ Links ]
11. Mills JA, Michel BA, Bloch DA y col. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheumatol 1990; 33 (8): 1114-1121. [ Links ]
12. Masuda M, Nakanishi K, Yoshizawa N, Iijima K y Yoshikawa N. Group A streptococcal antigen in the glomeruli of children with Henoch-Schönlein nephritis. Am J Kidney Dis 2003; 41: 366–370. [ Links ]
13. Yao-Hsu Yang a, Ya-Hui Chuang b, Li-Chieh Wang a, Hsin-Yi Huang a, M. Eric Gershwin c, Bor-Luen Chiang a. Department of Pediatrics. National Taiwan University Hospital. Autoimmunol Rev 2008; 7 (3): 179-843. [ Links ]
14. Gedalia A. Henoch-Schönlein purpura. Curr Rheumatol Rep 2004; 6 (3):195-202. [ Links ]
15. Saulsbury FT. Clinical update: Henoch-Schönlein purpura Lancet 2007; 369 (9566): 976-978. [ Links ]
16. Bailey M, Chapin W, Licht H y Reynolds JC. The effects of vasculitis on the gastrointestinal tract and liver. Gastroenterol Clin North Am 1998; 27 (4): 747-782. [ Links ]
17. Fervenza FC. Henoch-Schönlein purpura nephritis. Int J Dermatol 2003; 42 (3): 170-177. [ Links ]
18. Shin JI, Park JM, Shin YH, Hwang DH, Kim JH y Lee JS. Predictive factors for nephritis, relapse, and significant proteinuria in childhood Henoch-Schönlein purpura. Scand J Rheumatol 2006; 35 (1): 56-66. [ Links ]
19. Mc Carthy HJ y Tizard EJ. Clinical practice: Diagnosis and management of Henoch–Schönlein purpura. Eur J Pediatr 2009; 169: 643-650. [ Links ]
20. Gedalia A. Henoch-Schönlein purpura. Curr Rheumatol Rep 2004; 6 (3):195-202. [ Links ]
21. Zampetti A y col. Longitudinal study of microvascular involvement by nailfold capillaroscopy in children with Henoch–Schönlein purpura. Clin Rheumatol 2009; 28: 1101–1105. [ Links ]
22. Martino F, Agolini D, Tsalikova E y col. Nailfold capillaroscopy in Henoch-Schönlein purpura: a follow-up study of 31 cases. J Pediatr 2002; 141: 145. [ Links ]
23. Schwab J, Benya E, Lin R y Majd K. Contrast enema in children with Henoch-Schönlein purpura. J Pediatr Surg 2005; 40 (8): 1221-1223. [ Links ]
24. Ronkainen J, Koskimies O, Ala-Houhala M y col. Early prednisone therapy in Henoch-Schönlein purpura: a randomized, double-blind, placebo-controlled trial. J Pediatr2006; 149 (2): 241-244. [ Links ]
25. Weiss PF, Feinstein JA, Luan X, Burnham JM y Feudtner C. Effects of corticosteroid on Henoch-Schönlein purpura: a systematic review. Pediatrics 2007; 120 (5):1079-1087. [ Links ]
26. Zaffanello M, Brugnara M y Franchini M. Therapy for children with Henoch-Schönlein purpura nephritis: a systematic. Sci World J 2007; 7: 20-30. [ Links ]
27. Narchi H. Risk of long-term renal impairment and duration of follow up recommended for Henoch-Schönlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child 2005; 90 (9): 916-920. [ Links ]
28. Goldstein AR, White RH, Akuse R y Chantler C. Long-term follow-up of childhood Henoch–Schönlein nephritis. Lancet 1992; 339 (8788): 280–282. [ Links ]
29. Ronkainen J, Nutinen M y Koskimies O. The adult kidney 24 years after childhood Henoch–Schönlein purpura: a retrospective cohort study. Lancet 2002; 360 (9334): 666–670. [ Links ]

