










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista argentina de dermatología
On-line version ISSN 1851-300X
Rev. argent. dermatol. vol.95 no.1 Ciudad Autónoma de Buenos Aires Jan./Mar. 2014
CASOS CLÍNICOS Y REVISIÓN
Xeroderma pigmentoso. Breve revisión: de lo molecular a lo clínico
Xeroderma pigmentosum. Brief review: from molecular to clinical
AD Pérez-Elizondo *, GT del Pino-Rojas ** y JF García-Hernández ***
*Dermatooncólogo. Jefe de la Consulta Externa del Hospital para el Niño. Instituto Materno-Infantil del Estado de México. Profesor Titular de la Cátedra de Dermatología. Universidad Autónoma del Estado de México. Presidente de la Academia Universitaria de Dermatología.
** Dermatoncóloga. Práctica Privada.
*** Médico Residente de Primer año de Pediatría. Hospital para el Niño. Instituto Materno-Infantil del Estado de México.
Autor corresponsal: Antonio David Pérez-Elizondo.
Valladolid 3-903. Colonia Roma Norte. México D.F. Teléfono:- 52545298
E-mail: antoniodavid64@gmail.com / apederma@yahoo.com.mx
Recibido: 20.01.2014
Aceptado para su publicación: 25.02.2014
RESUMEN
El xeroderma pigmentoso es una rara genodermatosis transmitida de forma autosómica recesiva, de diagnóstico eminentemente clínico y pronóstico ominoso. Está condicionado por defectos moleculares intrínsecos en la reparación del DNA celular, dañado por efectos lumínico-radiantes con la consecuente acumulación de foto-productos oncogénicos e inhibición de genes supresores del crecimiento tumoral. Son evidentes las manifestaciones cutáneas, oculares y neurológicas degenerativas desde temprana edad. Representa un reto terapéutico y es indispensable la participación médica multidisciplinaria.
PALABRAS CLAVE: Xeroderma; Genodermatosis; Cáncer de piel.
SUMMARY
Xeroderma Pigmentosum (XP) is a rare autosomal recessive transmitted genodermatosis. The diagnostic is clinical and usually have a poor prognosis. There are intrinsic molecular defects in cellular DNA, repair damaged by radiant effects of the sun resulting in accumulation of oncogenic photoproducts and inhibition of tumor growth suppressor genes. It can produce serious affectation of the eye, central nervous system and skin. Represents a therapeutic challenge and is mandatory a multidisciplinary medical intervention for its management.
KEY WORDS: Xeroderma pigmentosum; Genodermatosis; Skin cancer.
INTRODUCCIÓN
Descrito originalmente por Kaposi en 1863, el xeroderma pigmentoso es una rara genodermatosis transmitida de forma autosómica recesiva. Se caracteriza por una temprana e intensa sensibilidad tegumentaria a la radiación lumínico-solar, cursando con quemaduras solares frecuentes, sequedad, discromías y cambios poiquilodérmicos con el desarrollo de neoplasias durante los primeros años de la niñez. Este trastorno progresivamente desfigurante y letal, está condicionado por anomalías en la escisión y reparación del DNA celular, dañado por el espectro ultravioleta de corta longitud de onda, entre los 280 y 320 nm (espectro UVB). A inicios de la década de los 30, de Sanctis y Cacchionne comunicaron alteraciones neurológicas degenerativas asociadas a la enfermedad, lo que contabiliza el 20-30% de todos los casos afectados. Cleaver en 1968, estableció el origen molecular al demostrar numerosos defectos en la reparación del DNA, en cultivos de fibroblastos irradiados con luz ultravioleta. Sin duda representa un desafío diagnóstico y terapéutico para el médico tratante; se aconseja la participación de varias especialidades entre las más importantes: pediatría, genética, dermatología, neurología y oftalmología para su adecuada intervención integral 1,2,3.
DATOS EPIDEMIOLÓGICOS
La incidencia mundial del padecimiento es de dos a cuatro nacidos vivos, por millón de habitantes, aunque varía según diferentes regiones geográficas, probablemente por razones étnico-genéticas; el índice de consanguinidad es alto. La frecuencia se calcula en los Estados Unidos de América y Europa de 1:250.000, mientras que en Japón asciende a 1:40.000. La tasa en los países latinoamericanos es más baja. No existe predilección por género. Bradford y col documentan recientemente, que existe un riesgo potencial de hasta 2.000 veces más para desarrollar melanoma y casi 10.000, para carcinomas basocelulares y espinocelulares, en pacientes con xeroderma pigmentoso comparado con la población general. Se estima además un aumento en el desarrollo de neoplasias malignas de hasta veinte veces más, involucrando cerebro, pulmones, tracto gastrointestinal y riñones 4,5,6.
ETIOPATOGÉNESIS
La fracción ultravioleta B del espectro fotobiológico, es absorbida por cromóforos cutáneos como el ADN celular, dañando su estructura molecular formando dos fotoproductos: dímeros de ciclobutano timina y dímeros de citosina y timina. Estas moléculas pueden interferir en la transcripción y replicación normales, de la conformación helicoidal del DNA distorsionado. Un complejo proceso de reparación molecular, que relaciona la participación de al menos 30 proteínas denominado REN, escisión selectiva de nucléotidos, implica la acción del sistema global de corrección del genoma y la reparación acoplada a la transcripción (Fig 1).
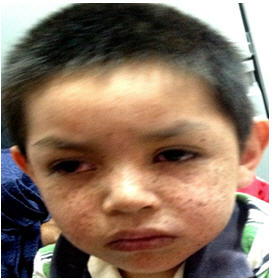
Fig 1: etapa poiquilodérmica temprana
Consiste en seis pasos fundamentales:
1) Reconocimiento y localización lesional / XPA, XPC y XPE.
2) Desenrollamiento de la doble hélice del DNA / XPB y XPD.
3) Actividad endonucleasa / XPF y XPG.
4) Acción exonucleasa con eliminación del nucléotido dañado.
5) DNA polimerasa con neosíntesis de nucléotidos.
6) DNA ligasa con unión de la hebras contiguas complementarias.
Los efectos nocivos del metabolismo tisular oxidativo, exposición química tóxica y particularmente la radiación ionizante y la fracción B de la luz ultravioleta, en pacientes con capacidad reducida o ausente, para la reparación del material genómico y pérdida de la vigilancia de la estructura y función del mismo, conduce al acúmulo aberrante de mutaciones oncogénicas e inhibición de genes supresores de crecimiento tumoral; lo anterior se ejemplifica en la Tabla I.
TABLA I. Grupos de complementación, genes implicados y procesos afectados en el XP
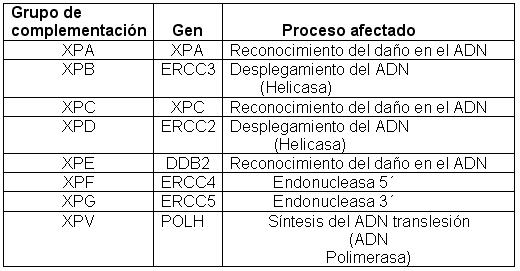
La gravedad, extensión e intensidad de los datos clínicos se relaciona con la naturaleza del defecto de los genes, que codifican para diferentes enzimas de reparación molecular. Así, se conocen siete grupos de complementación y una variante en la que no hay afectación neurológica, detalles que se muestran en las Tablas II. 7,8,9,10.
TABLA II. Frecuencia de los grupos de complementación en el XP

CUADRO CLÍNICO
Las primeras manifestaciones de la enfermedad, se observan entre el sexto mes y tercer año de vida en el 75% de los casos; afecta las partes corporales foto-expuestas tales como: la cara, pabellones auriculares, cuello, V del escote, aspectos externos de las extremidades, dorso de manos, así como conjuntivas y labios. Se identifican tres etapas evolutivas que bien pueden sobreponerse 11,12,13,14:
1) Fase Eritemato Pigmentaria:
Existe eritema moderado a intenso, a veces con edema difuso y aparición de vesico-ampollas, tras una exposición solar mínima durante la lactancia, lesiones cutáneas que van haciéndose más frecuentes e intensas con el transcurso de la edad; hallazgo observado hasta en el 50% de los afectados. En los primeros años de la vida son evidentes pequeñas máculas idénticas a efélides, lenticulares, de color marrón más o menos intenso, agrupadas y con tendencia a confluir, junto a éstas aparecen manchas acrómicas irregulares. El paciente experimenta fotofobia y cuadros de queratoconjuntivitis de repetición.
2) Fase Atrófico Telangiectásica:
Gradualmente se desarrollan zonas atrófico-nacaradas dispersas con numerosas telangiectasias superficiales y cicatrices; la piel está seca, escamosa y corrugada, condición denominada poiquilodermia. El progresivo adelgazamiento y perfilamiento de la nariz, microstomía, mutilación auricular; ectropión deformante, madarosis, opacidades corneales y los pterigion son datos fenotípicos sugestivos (Fig 1).
3) Fase de Proliferación Cutánea Tumoral:
En la etapa escolar comienzan a desarrollarse formaciones verrugosas blanquecinas, cuernos cutáneos, queratosis actínicas (carcinomas espinocelulares muy superficiales) y angiofibromas. Se establecen neoplasias mucocutáneas malignas como carcinomas basocelulares y espinocelulares infiltrantes, melanomas y sarcomas muchas veces agresivos, infiltrantes y destructivos (Fig 2).
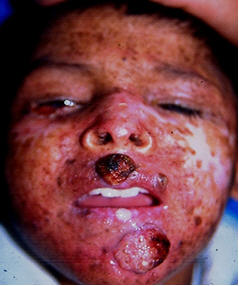
Fig 2: fase atrófica-tumoral.
La afección ocular es casi tan frecuente como las manifestaciones tegumentarias, apareciendo hacia los cuatro años de vida; los hallazgos más notorios son queratoconjuntivitis crónica, vascularización y opacidades corneales, pterigion y pinguécula. El adelgazamiento extremo de la piel palpebral resulta en ectropión, endoprión y el desarrollo de carcinomas o melanoma.
Alrededor del 20-30% de los casos con xeroderma pigmentoso, tienen una enfermedad neurológica degenerativa asociada de curso e intensidad muy variables. Se puede manifestar desde la infancia temprana o entrada la adolescencia; son hallazgos comunes: la microcefalia, retraso en el desarrollo psicomotor, epilepsia, ataxia, espasticidad, arreflexia y pérdida auditiva.
El síndrome de de Sanctis-Cacchionne se reserva a los casos de xeroderma pigmentoso acompañados de: enanismo, inmadurez sexo-genital y grave deterioro neurológico15,16.
DIAGNÓSTICO
El reconocimiento distributivo y morfológico de las lesiones nos orienta al diagnóstico de xeroderma pigmentoso; la biopsia cutánea con anestesia local de una neoformación sospechosa, nos confirma la naturaleza y origen de una neoplasia maligna y su oportuna extirpación. Se puede realizar diagnóstico prenatal en padres portadores por amniocentesis, así como diversas pruebas ultraestructurales en laboratorio especializado de citogenética, para determinar la hipersensibilidad celular, anomalías cromosómicas y defectos en la reparación del DNA, métodos confirmatorios muchas veces fuera del alcance nosocomial 17,18,19.
DIAGNÓSTICO DIFERENCIAL
La fase eritematosa temprana que simula una quemadura solar, debe distinguirse de una reacción de fotosensibilidad por fármacos, protoporfiria eritropoyética, síndrome de Rothmund-Thompson o enfermedad de Hartnup. En etapas tardías se descartarían cuadros esclerodermiformes, urticaria pigmentosa, epidermodisplasia verruciforme, síndrome de Cockayne o tricodistrofia 20.
TRATAMIENTO
El diagnóstico oportuno y la protección rigurosa frente a las radiaciones lumínico-solares, constituyen la piedra angular en el manejo de los casos afectados. Es imprescindible educar a los familiares responsables y al propio paciente, sobre los efectos dañinos de la luz solar y otras fuentes radiantes como el neón y halógeno; el empleo frecuente y constante de ropa apropiada, gafas, sombreros y sombrillas como medidas físicas de protección. La aplicación de filtros y pantallas antisolares cada 2-4 horas en piel fotoexpuesta, debe ser la regla.
Es aconsejable evaluar los riesgos y beneficios de medicamentos de uso común, potencialmente foto-alérgicos o fototóxicos, tales como: antimicrobianos, antiinflamatorios no esteroideos, diuréticos, anticonvulsivantes o antihipertensivos, entre otros.
Algunos autores aconsejan la administración de productos antioxidantes como las vitaminas A, C, E y especialmente la D o dieta rica en éstos, para evitar cuadros carenciales.
Los derivados sistémicos del ácido retinoico, isotretinoína o acitretina, parecen ser eficaces en la prevención de tumores malignos, actúan mientras son utilizados requiriendo dosis altas y largo tiempo de administración, no están exentos de efectos secundarios.
En caso de queratosis actínicas y carcinomas superficiales son efectivos: el 5-fluorouracilo en crema al 5% untado por las noches durante períodos de 1-2 meses, con la esperada irritación cutánea y/o un inmunomodulador, como el imiquimod en carcinomas basocelulares poco agresivos y situados en áreas de bajo riesgo. Sin duda representa un avance actual en el tratamiento de esta afección.
Las formaciones neoplásicas pre malignas y malignas pueden ser intervenidas mediante electro desecación y curetaje, crioterapia con nitrógeno líquido, resección quirúrgica simple con márgenes amplios, colgajos o injertos según la lesión lo amerite (Figs 5 y 6). En nuestro Servicio de Dermatología ocasionalmente son de utilidad: las quimio-exfoliaciones de profundidad media con ácido tricloroacético al 35% o las dermoabrasiones mecánicas, bajo anestesia local y sedación (Figs 3 y 4) 21,22.

Figs 3 y 4: quimio-exfoliaciones con ácido tricloroacético (2), antes y después.

Figs 5 y 6: extirpación de carcinoma espinocelular de la punta nasal.
CONCLUSIÓN
El xeroderma pigmentoso es una genodermatosis de transmisión autosómica recesiva, caracterizada por hipersensibilidad a la radiación solar cursando con episodios iniciales de quemadura solar, discromía óculotegumentaria, cambios poiquilodérmicos, así como rápida y temprana aparición de tumores de pronóstico ominoso. Se determina una incapacidad enzimática natural para eliminar fotoproductos dañinos para el DNA celular. Es recomendable la asesoría genética y la participación médica multidisciplinaria, para su evaluación periódica de control.
1. Ortellao L, Rambaldo L. Xeroderma pigmentoso: presentación de 2 casos. Arch Argent Pediatr 2007; 105 (5): 432-435. [ Links ]
2. Spitz JI. Genodermatosis. A clinical guide to genetic skin disorders. Segunda Edición. Lippincott. Williams & Wilkins. Philadelphia 2005; 174-177. [ Links ]
3. Guillén Sánchez-Pedreño P, Martínez Liarte J. Xeroderma Pigmentoso. Piel 2004; 19 (7): 364-373. [ Links ]
4. Chantorn R, Lim HW, Shwayder TA. Photosensitivity disorders in children. Part II. J Am Acad Dermatol 2012; 67: 1113-1127. [ Links ]
5. Bradford PT, Goldstein AM, Tamura D y col. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterizes the role of DNA repair. J Med Genet 2011; 48: 168-176. [ Links ]
6. Kraemer KH, Tamura D, Khan SG, DiGiovanna JJ. Burning issues in the diagnosis of xeroderma pigmentosum. Br J Dermatol 2013; 169:1175-1179. [ Links ]
7. Díaz Leonard D, Herrera Alonso A, Vigera Fajardo M. Xeroderma Pigmentoso. Presentación de un caso. Medi Sur 2008; 6 (2): 97-100. [ Links ]
8. Mena Cedillo C, Arroyo Pineda A. Xeroderma Pigmentoso. Bol Med Hospital Infantil de México 1996; 53 (4): 192-196. [ Links ]
9. Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS. Patología Estructural y Funcional. Séptima Edición. Elsevier Saunders Editorial. 2005; 311-327. [ Links ]
10. Saúl A. Lecciones de Dermatología. Méndez Editores. Décimo Quinta Edición. 2008; 655-686. [ Links ]
11. Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, Mc Kenna WG. Oncología Clínica. Volumen I. Tercera Edición. Elsevier Churchill Livingstone Editorial. 2005; 193-198. [ Links ]
12. Nussbaum RL, McInnes RR, Willard HF. Genética en Medicina. Thompson & Thompson. Séptima Edición. Elsevier Masson Editorial. 2008; 318-319. [ Links ]
13. Lehmann AR, McGibbon D, Stefanini M. Xeroderma Pigmentosum. Orphanet J Rare Dis 2011; 6:70. [ Links ]
14. Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A, Armier J, Pham D, Khadir K, Roume J, Hadj-Rabia S, Bouadjar B, Taieb A, Verneuil H, Benchiki H, Grandchamp B, Sarasin A. A Prevalent Mutation with Founder Effect in Xeroderma Pigmentosum Group C from North Africa. J Invest Dermatol 2010; 130: 1537-1542. [ Links ]
15. Liy WM, Durán McKinster C, Orozco CL, Sáez OM, Carrasco DD, Ruiz Maldonado R. Xeroderma pigmentoso con retraso psicomotor: Síndrome de De Sanctis Cacchione. Reporte de dos casos de origen mexicano. Dermatol Pediatr Lat 2004; 2 (1): 50-53. [ Links ]
16. Falcón Lincheta L, Dorticós Balea A, Simón RD, Garbayo Otaño E. Xeroderma Pigmentoso. Síndrome de Sanctis Cacchione. Presentación de un caso. Rev Cubana Pediatr 1998; 70 (2): 113-116. [ Links ]
17. Camargo R, Choque Pardo J, Magne Rojas WS, Madariaga Bedoya JN. Xeroderma Pigmentoso. Rev Bol Pediatr 2008; 47 (1): 16-18. [ Links ]
18. Rodríguez-García R, Aguilar-Ye A, Puig Sosa PJ, Solis-Daun O, Padilla-Castillo A. Xeroderma pigmentoso en dos hermanas. Rev Mex Pediatr 2002; 69 (4): 151-154. [ Links ]
19. Arenas R. Dermatología. Atlas, diagnóstico y tratamiento. Cuarta Edición. Mc Graw Hill Interamericana. México 2009; 369-371. [ Links ]
20. Kleijer WJ, Laugel V, Berneburg M y col. Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008; 7: 744-750. [ Links ]
21. Butt FM, Moshi JR, Owibingire S, Chindia ML. Xeroderma pigmentosum: a review and case series. J Craniomaxillofac Surg 2010; 38: 534-537. [ Links ]
22. Eichenfield L, Álvarez Connelly E. Máculas hiperpigmentadas. En: Pueyo de Casabé S y Valverde R. Dermatología Neonatal. Buenos Aires. Artes Gráficas Buschi. 2005; 211-212. [ Links ]

