










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista argentina de dermatología
On-line version ISSN 1851-300X
Rev. argent. dermatol. vol.98 no.4 Ciudad Autónoma de Buenos Aires Dec. 2017
CASO CLÍNICO PATOLÓGICO
Síndrome de bart. A propósito de un caso
Bart syndrome. Report of a case
AD Pérez- Elizondo * y A Valdés-López **
*Dermato-Oncólogo. Jefe de la Consulta Externa del Hospital para el Niño. Instituto Materno-Infantil del Estado del México. Universidad Autónoma del Estado de México. Presidente de la Academia Universitaria de Dermatología. Academia Mexicana de Pediatría.
** Médico Neonatólogo. Jefe del Servicio de Neonatología. Hospital para el Niño. Instituto Materno-Infantil del Estado de México.
e-mail:antoniodavid64@gmail.com
Los autores declaramos no poseer ningún tipo de conflicto de interés.
Recibido: 21.08.2017. Aceptado para su Publicación: 05.10.2017.
RESUMEN
El síndrome de Bart es un trastorno congénito poco frecuente, caracterizado por la asociación de epidermólisis ampollosa, ausencia congénita localizada de piel y ocasionalmente anormalidades ungueales. En este artículo se reporta el caso de un neonato masculino, remitido al Hospital para el Niño del IMIEM (Instituto Materno-Infantil del Estado de México), para valoración de lesiones ampollosas extensas y ausencia de piel en miembros inferiores, presentes desde el nacimiento quien después del tratamiento, mostró mejoría con una evolución clínica favorable.
PALABRAS CLAVE: síndrome de Bart, aplasia cutis, epidermólisis ampollosa.
SUMMARY
Bart syndrome is a rare congenital disorder characterized by the association of epidermolysis bullosa, localized congenital absence of skin and occasionally nail abnormalities. In this presentation we report the case of a male neonate referred to the Hospital para el Niño of IMIEM for evaluation of extensive blistering lesions and absence of skin in lower limbs, present from birth and who after treatment showed improvement, with a favorable clinical evolution.
KEY WORDS: Bart syndrome, aplasia cutis, epidermolysis bullosa.
CONCEPTO
Bart y col en 1966, describieron el caso de una familia que presentaba ausencia congénita localizada de piel, además de lesiones vesicoampollosas mucotegumentarias y alteraciones ungueales, con un patrón de herencia autosómica dominante y penetrancia incompleta, de variable expresión fenotípica. Posteriormente, varios autores concluyen que corresponde a un subtipo muy particular o variante alélica, de la epidermólisis ampollosa distrófica dominante, acompañada de aplasta cutis congénita. Se ha demostrado una disminución de las fibrillas de anclaje y la separación se sitúa por debajo de la lámina densa; de igual forma se detecta que esta compleja enfermedad, está relacionada con mutaciones genéticas en el brazo corto del cromosoma 3 (3p21), que codifica el colágeno tipo VII (COL7A1). Clínicamente, la aplasia cutis es bilateral y simétrica afectando extremidades inferiores, particularmente en la región pretibial siguiendo las líneas de fusión embrionarias; hay reportes de involucro unilateral. En el período neonatal, aparecen las lesiones vesicoampollosas en áreas corporales sometidas a traumatismos friccionales; además, es frecuente observar manchas discrómicas, quistes de inclusión y cicatrices postlesionales 1,2,3.
PRESENTACION DEL CASO
Se interconsulta al Servicio de Dermatología del Hospital por neonato de 15 días de vida, producto de término con peso y talla dentro de percentilas normales, sin sintomatología general alguna. Se evidencian a la exploración física detallada, placas erosivoulceradas de base eritematosa húmeda granular, irregulares, bien definidas, cubiertas por una fina membrana blanquecino-grisácea fácilmente desprendible, que deforman y dificultan el movimiento de ambos pies; se extienden desde la porción distal de la región tibial, dorso y parte de los dedos. Hallazgo compatible con aplasia cutis congénita bilateral y simétrica (Figs 1 y 2). En tronco y miembros superiores particularmente en zonas acrales, se observan numerosas formaciones vesicoampollares de contenido seroso y paredes adelgazadas, así como zonas erosivocostrosas circulares, ovaladas, de tamaños diferentes y desprendimientos laminares de escama, correspondientes a las ampollas rotas dejando áreas cutáneas denudadas (Figs 3, 4 y 5). No hay antecedentes perinatales ni personales patológicos de importancia, para el padecimiento actual. Se protocoliza el caso solicitando biometría hemática completa, tiempos de coagulación, química sanguínea, exudado faríngeo y examen general de orina, reportándose sin alteraciones.
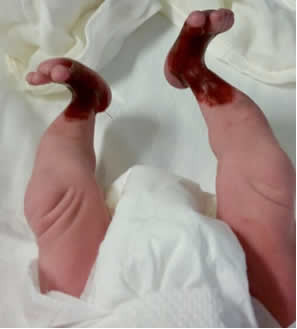
Fig 1: ausencia congénita de piel en ambos pies.

Fig 2: detalle lesional.
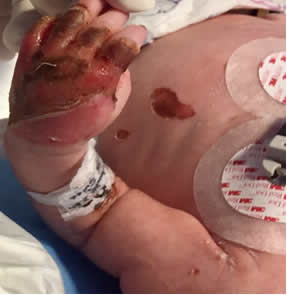
Fig 3: erosiones, costras y descamación
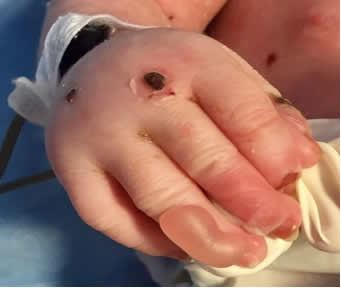
Fig 4: ampollas y erosiones cotrosas en mano.
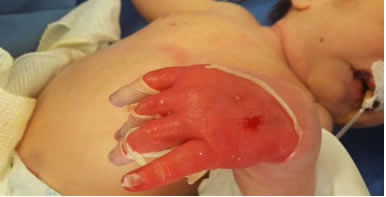
Fig 5: piel denudada con descamación laminar en mano.
Frente a la sospecha diagnóstica, se efectúa biopsia lesional para estudio histopatológico. Se encuentran algunos queratinocitos basales desprendidos y hendiduras ampollares subepiteliales, sin infiltrado inflamatorio dérmico compatible con epidermólisis ampollosa (Fig 6).

Fig 6: ampolla subepidérmica (H&E 40x).
Se practica aseo aséptico con solución de clorhexidina y aplicación de gasas estériles, impregnadas con crema de manzanilla y mupirocina sobre las áreas erosionadas, cada 12 horas. Del mismo modo, durante su estancia hospitalaria a nivel de los pies se colocan parches hidrocoloides oclusivos, con cambio cada cinco días por cuatro meses hasta la aparición de una escara, cuya caída deja una cicatriz amarillento-nacarada atrófica. En las visitas médicas de control se evidencia disminución de la frecuencia, extensión e intensidad en el desarrollo de ampollas, así como mejoría de la apariencia física y funcional de las cicatrices de los miembros inferiores. Después de dos años de vigilancia periódica se le da de alta.
COMENTARIO
El síndrome de Bart corresponde a una rara enfermedad, que se asocia a una aplasia cutis congénita muchas veces bilateral y simétrica, involucrando principalmente a los miembros inferiores, deformidades ungueales y epidermólisis ampollosa. Aunque de origen incierto, se atribuye a mutaciones puntuales del cromosoma 3p. Descrita inicialmente por Cordon en 1767, la aplasia cutis es un defecto tegumentario que puede extenderse hasta fascia, músculos e incluso hueso. La ausencia congénita de piel de localización preferente en la piel cabelluda, es referida por primera vez por Campbell en 1826; de hecho es la topografía más común en la práctica diaria.
Frieden clasificó la aplasia cutis congénita en nueve grupos, de acuerdo con: la localización topográfica, factores hereditarios y anomalías asociadas; así el síndrome de Bart correspondería al tipo VIa con afectación de extremidades, incluso desde la rodilla y extensión serpiginosa hasta el cuello del pie, abarcando dorso y dedos. Pueden aparecer uñas distróficas y metatarso varo, aunque no siempre se presentan todos los datos referidos, tal como ocurre con el caso expuesto.
Sin duda el diagnóstico de este raro padecimiento es básicamente clínico, la presencia de formaciones ampollosas subepidérmicas con separación de la lámina subdensa, representa un apoyo confirmatorio. El manejo es siempre conservador a base de medidas que eviten los traumatismos accidentales y prevención de infecciones secundarias, mediante el empleo de antibióticos tópicos. Por lo regular, el pronóstico es favorable con adecuado crecimiento del niño, reepitelización con secuelas cicatrizales a los pocos meses y la tendencia a desaparecer los brotes ampollosos espontáneos o traumáticos, después de la pubertad 4,5,6.
El interés de presentar este caso, se fundamenta en la singularidad de su presentación en la práctica cotidiana del médico, poder identificar e interrelacionar los hallazgos tegumentarios y en lo posible, manejarlo de manera multidisciplinaria.
1. Aygun AD, Yilmaz E, Kurt AN, Kurt A, Elkiran O, Okur I y Ozercan I. Aplasia cutis congenita and epidemolysis bullosa: Bart syndrome. Int J Dermatol 2010; 49: 343-345.
2. McCarthy MA, Clarke T y Powell FC. Epidermolysis bullosa and aplasia cutis. Int J Dermatol 1991; 30: 481-484. [ Links ]
3. Prada J y Rojas L. Síndrome de Bart. Reporte de un caso. Rev Med 2008; 16: 232-236. [ Links ]
4. Siáñez-González C, Pezoa-Jares R y Salas-Alanis JC. Congenital epidermolysis bullosa: a review. Actas Dermosifiliogr 2009; 100: 842-856. [ Links ]
5. Frieden IJ. Aplasia cutis congenital: a clinical review and proposal for classification. J Am Acad Dermatol 1986; 14: 646-660. [ Links ]
6. Boente MC, Asial RA, Del Valle Frontini M, Primc NB y Winik BC. Epidermólisis Ampollar con Ausencia Congénita Localizada de piel (Síndrome de Bart). ¿Es posible hablar de pérdida de heterozigocidad? Dermatol Pediatr Lat 2004; 2 (2): 125-129. [ Links ]

