











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las neurofibromatosis son un grupo de enfermedades genéticas multisistémicas, heredadas de forma autosómica dominante con implicación patogénica de: la piel, sistema nervioso, ojos, huesos y sistema endócrino, con un amplio espectro de: hamartomas, tumores malignos y alteraciones congénitas, incluidas dentro de las llamadas facomatosis. Las clasificaciones han sido variadas, desde los 7 tipos de Riccardi (clásica, acústica, mixta, inclasificable, segmentaria, sólo manchas «café con leche», sólo neurofibromas), pasando por las 4 formas de Houson (clásica, acústica, segmentaria, manchas «café con leche» familiares) hasta la clasificación actualmente más utilizada mediante la que, sólo se consideran 2 tipos: neurofibromatosis tipo 1 (NF-1) y neurofibromatosis tipo 2 (NF-2) con una variante de NF-1 que sería la neurofibromatosis segmentaria.1
La neurofibromatosis tipo 1 (NF1), también llamada periférica o enfermedad de von Recklinghausen, en honor al patólogo alemán Friedrich von Recklinghausen (1833-1910), quien la describió por primera vez en1884 2, es el más frecuente de los síndromes neurocutáneos (1:2000 a 2500 recién nacidos), siendo responsable del 85% de todo los casos de neurofibromatosis. 3
Su herencia es autosómica dominante causada por un espectro de mutaciones del gen NF-1, aunque hasta en el 50% de los casos se debe a mutaciones espontáneas (mutaciones de novo). El riesgo de desarrollar la NF-1 mediante una neo-mutación en un individuo, está en relación con la edad paterna avanzada. Por este motivo más del 90 % de las mutaciones esporádicas, procede de alelos heredados del padre. 4
Causado por una mutación del gen NF1 que codifica la neurofibromina, se trata de un gen regulador negativo de la vía ras/mitogen-activated protein kinasa.5
La expresión clínica de la NF1 es variable. Desde pequeños neurofibromas dérmicos y pigmentación anormal, hasta grotescos neurofibomas plexiformes y displasias óseas, que interfieren con la función de órganos o alteran la apariencia del individuo. Su manifestación clínica más importante es la aparición de lesiones cutáneas (máculas café con leche), que pueden estar en cantidades de 500 a 1.000, principalmente en tórax. 5
En 1987, el National Institute of Health (NIH) Consensus Development Conference estableció los siete criterios diagnósticos de la enfermedad, que se revisan periódicamente:
Maculas café con leche (MCCL) (seis o más, menores de 5 mm en pacientes prepúberes y mayores de 15 mm, en pospuberales).
Neurofibromas (dos o más de cualquier tipo, uno solo si es plexiforme).
Lentiginosis axilar (signo de Crowe).
Glioma óptico.
Nódulos de Lisch o hamartomas pigmentarios del iris (dos o más).
Displasia del esfenoides o adelgazamiento de los huesos largos, con seudo artrosis o no.(tibia en sable).
Antecedentes familiares de primer grado.
El diagnóstico requiere la presencia de al menos dos de estos criterios, los que tienen alta sensibilidad y especificidad en la edad adulta, no así en la infancia.5 Es por ello, que en los últimos años se han tratado de definir otros hallazgos clínicos de aparición temprana, a fin de contribuir al diagnóstico precoz de la enfermedad.
En el año 2000 De Bella K y col concluyeron en un estudio, aplicado a más de 1.900 pacientes con NF1, que el diagnóstico no siempre se puede hacer en niños pequeños usando los criterios de diagnósticos. En dicho estudio se demostró, que el 46% de los casos esporádicos no cumplía con los criterios suficientes, para establecer el diagnóstico antes del año de edad; sin embargo, el 97% los presentaba a la edad de 8 años y el resto, los completaba hacia los 20 años, concluyendo que la modificación de estos criterios puede ser necesaria, para niños menores de 8 años. 6
A pesar del descubrimiento de la mutación genética, que se localiza en el brazo largo del cromosoma 17 (17q) y de la identificación de la proteína, involucrada en la producción de la enfermedad (neurofibromina), el sustrato fundamental del diagnóstico sigue estando en las alteraciones clínicas.
La NF1 es un síndrome heterogéneo, ya que, prácticamenteno existe una sola zona del cuerpo que queda libre de sufrir algún tipo de alteración y progresiva pues, aunque sus manifestaciones más características pueden estar presentes al nacer, la aparición de complicaciones puede retrasarse. Las manchas café con leche aparecen casi en el 100% de los pacientes. Otras manifestaciones se ven con menos frecuencia: alteraciones óseas (fundamentalmente de columna, en forma de escoliosis o cifoscoliosis); oculares (las más frecuentes son los nódulos de Lisch, hamartomas benignos del iris presentes en el 90% de los casos); endocrinológicas (talla corta, pubertad precoz); fenotipo Noonan; hemihipertrofia; problemas neuropsicológicos; anomalías vasculares y patología del sistema nervioso central como: crisis epilépticas, siringomielia, meningoceles radiculares y estenosis del acueducto de Silvio; esta última, una de las complicaciones más habituales y una de las principales. 7
CASO CLÍNICO
Se trata de un escolar masculino de 8 años de edad, natural y procedente de Valencia Estado Carabobo Venezuela. Fototipo de piel: IV / VI (según Fitzpatrick). Sin antecedentes personales de interés, quien presenta dermatosis generalizada bilateral y asimétrica, a predominio de tórax y región inguinal caracterizada por maculas café con leche, en número mayor de 6 de diferentes tamaños; en tórax anterior se aprecia mácula que abarca desde línea axilar posterior derecha, hasta la línea media clavicular izquierda, de 30 x 40 cm de longitud dentro de la que presenta zona pardo oscura desde su nacimiento, con pelos en su superficie de un año de evolución (Figs 1,2). Asimismo en región inguinal mácula bien definida, con efelides satélites, signo de Crowen positivo también desde su nacimiento (Fig 3).
Evidenciamos deformidad ósea, caracterizada por: aumento del diámetro ántero posterior del cráneo, desviación de la cintura escapular derecha, cifoescoliosis y baja estatura proporcionada (Figs 4,5).
Entre los antecedentes familiares se encuentra: madre con maculas café con leche y múltiples neurofibromas en piel.
Se plantean como diagnósticos presuntivos: neurofibromatosis, síndrome del nevo de Becker, síndrome de McCune - Albright y síndrome de Legius.
Se realizan laboratorios de rutina los que no muestran alteraciones.
En vista de la múltiple expresión clínica se solicita estudio del paciente de forma multidisciplinaria.
1) Oftalmología: se evidenció la presencia de nódulos de Lisch.
2) Otorrinolaringología: sin alteraciones al momento de la evaluación.
3) Traumatología: se evidenció la presencia de cifoescoliosis derecha, cráneo dolicocéfalo, corta estatura proporcionada y osteopenia.
5) Asesoría genética: PEDIGREE - NF1: genealogía con dos casos segregantes de NF1 (Gráfico I).
Se concluye con un diagnóstico definitivo: neurofibromatosis tipo 1. Enfermedad de von Recklinghausen.
DISCUSIÓN
La NF-1 es un desorden genético hereditario, que se transmite bajo un patrón autosómico dominante, con penetrancia casi del 100% y variable expresividad fenotípica e incidencia de 1 en 2.500 a 3.000 individuos, lo que la hace una de las anomalías congénitas más comunes. 8
Es una enfermedad multisistémica ocasionada por mutaciones heterogéneas en el gen NF1, las que son esporádicas en aproximadamente el 50% de los casos. NF1 fue identificado y su proteína caracterizada por: Cawthon y col y Wallace y col, respectivamente, en 1990. La secuencia del ADNc fue descrita por Gutmann y Collins y Viskochil y col, en 1993. 9
El gen afectado se localiza en el cromosoma 17q11.2 (brazo largo del cromosoma 17) y pseudogenes en los cromosomas 2, 14, 15, 18, 21 y 22, cuyo producto final es una proteína llamada neurofibromina, con una acción tumoral supresora, responsable de la expresión dérmica y expresión ubicua en: fibroblastos de piel, cerebro, bazo, pulmón, músculo, neuroblastoma, neurofibroma, células de neurofibrosarcoma, carcinoma de colon y cáncer de mama, entre otros.8
La neurofibromina se considera un supresor tumoral, pues la reducción de su producción o la completa pérdida de ésta, conlleva a la activación de la proteína ras, que a su vez regula una cascada de vías de señalización posteriores, entre ellas: las proteínas quinasas activadas por mitógenos, fosfoinositol 3-quinasas, proteína quinasa B y proteína quinasa diana de rapamicina en células de mamífero; la activación de estas vías, ocasiona una variedad de efectos celulares que, generalmente, estimulan la supervivencia y proliferación celular. 10,11
Sólo dos correlaciones genotipo-fenotipo, las que también, son ejemplos de la variabilidad fenotípica de NF1 han sido observadas: 1) la deleción completa del gen, micro deleciones NF1, caracterizada por la aparición temprana y numerosa de neurofibromas cutáneos, con mayor frecuencia y la severidad de anormalidades cognitivas, malformaciones, tumoración y riesgo aumentado de malignización y 2) la deleción específica de tres pb en el exón 17, c.2970-2972 delAAT (p.990delM), asociada con pigmentación, nódulos de Lisch y ausencia de neurofibromas cutáneos o neurofibromas plexiformes. Las mutaciones esporádicas en NF1 de la línea germinal, están sesgadas hacia el sexo. De éstas, más del 80% tienen un origen paterno. Otras mutaciones, llamadas micro deleciones del locus, tienen en su mayoría un origen materno.8
El Instituto Nacional de la Salud de EE.UU (NIH) definió en 1987, los criterios diagnósticos de NF1. El diagnóstico se basa en la valoración clínica y precisa de dos o más de los siete criterios establecidos.7

Siendo más comunes las máculas café con leche, presentes en el 99% de menores de 1 año, hamartomas melanocíticos del iris y efélides en aéreas no fotoexpuestas y los neurofibromas que se consideran casi patognomónicos de NF1.
Las máculas café con leche, representan acumulaciones de melanocitos generalmente de forma ovalada y son la manifestación inicial más frecuente. Estas pueden ser congénitas y su extensión y cantidad no se relacionan con la gravedad de la NF1, ni predicen la localización de los neurofibromas. Las máculas menores de 5 mm de diámetro se denominan “pecas o efélides” y se localizan sobre todo en axilas e ingles y permiten hacer el diagnóstico de NF1 en un niño pequeño, cuya única otra manifestación son las máculas café con leche. En general, aparecen entre los 3 y 5 años de edad. Si se encuentran en región submamaria constituyen el “signo de Crowe”, muy orientador en el diagnóstico. 2
En el caso de nuestro paciente la presencia de más de 6 máculas café con leche, al igual que efélides satélites y signo de Crowe positivo, nos permite realizar un diagnóstico clínico bastante certero.
Los neurofibromas son tumoraciones benignas derivadas de la vaina nerviosa, conformadas por células de Schwann, mastocitos, fibroblastos y células perineurales. Su consistencia es de suave a firme y el color es uniforme en diversas tonalidades de pigmentación, del rosado al café. Se localizan en piel, tejidos subcutáneos (se confunden con ganglios linfáticos), vísceras, sistema nervioso central y suelen aparecer en la pubertad. 2
Los neurofibromas plexiformes son más grandes, pueden estar asociados a hipertricosis y tener consistencia de “lombrices en su interior”, determinada por los trayectos nerviosos. En la histología son similares a neurofibromas; este tipo suele tener graves implicaciones estructurales y funcionales según su localización, por ejemplo, en genitales.12 Su crecimiento es caprichoso y en su interior se pueden gestar tumores malignos de la vaina nerviosa, muchas veces no detectados hasta que hay manifestaciones de sus metástasis, por lo que se recomienda un umbral bajo para biopsia ante el crecimiento, cambios clínicos y dolor. Histológicamente están conformados por células ahusadas con núcleos ondulados y fibrosis; también se encuentran mastocitos, fibroblastos, células perineurales y mucina. 2 El patrón de crecimiento es impredecible, con períodos de crecimiento sostenidos seguidos de crecimientos precipitados, la influencia hormonal puede modular su crecimiento, ya que, habitualmente muestran aumento de su tamaño durante la adolescencia y el embarazo. Varían considerablemente en número entre las familias, el síntomas más significativo que se les asocia es el prurito, siendo raramente dolorosos.
Aproximadamente el 15% desarrolla tumores de la vía óptica, de los que sólo la mitad presenta síntomas. La relación mujer/hombre es de 2:1 y se cree que hay factores hormonales en su génesis. El período de mayor riesgo es durante los primeros seis años de vida, por lo que es fundamental una exploración neurológica completa, siempre referir toda NF1 a evaluación neuro-oftalmológica y tener un alto nivel de sospecha ante datos de pubertad precoz, pues no es infrecuente encontrar un tumor en el quiasma. 2
Los nódulos de Lisch son hamartomas melanocíticos bilaterales benignos del iris, con elevaciones nodulares que se proyectan en la superficie del iris, variando su tamaño desde apenas visible hasta los 2 mm de diámetro, en la mayoría son de color café amarillento, pero a veces, particularmente en los niños, son muy pálidos incluso blancos. En algunos pacientes pueden ser visibles a ojo desnudo. Son patognomónicos de la NF1, no está claro si su incidencia aumenta con la edad. Para su diagnóstico se requiere el examen con lámpara de hendidura, por parte de un oftalmólogo familiarizado con la NF1.13
En el caso de nuestro paciente, a pesar de la sospecha de pubertad precoz en vista del desarrollo de genitales, no acorde a la edad de la evaluación neuro-oftalmológica, resultó dentro de los límites normales.
En cuanto a las lesiones esqueléticas afectan principalmente el ala de esfenoides (displasia), generando malformación en pared/piso de la órbita que puede pasar clínicamente inadvertida o provocar exoftalmos o endoftalmos; las vértebras y la tibia son el sitio de afección más común, lo que da lugar a problemas ortopédicos. La severidad es variable, generalmente progresiva y resistente a los tratamientos, siendo frecuente que existan neurofibromas para vertebrales y una densidad ósea disminuida. Generalmente el tratamiento de la escoliosis severa en la NF1 es insatisfactorio, la morbilidad y complicaciones post tratamiento no son frecuentes. 14
La escoliosis representa la manifestación esquelética más frecuente de NF1 (10 a 30%). Existen dos variedades: distrófica y no distrófica (la primera se presenta a edades más tempranas es la más grave, la segunda es más común en la adolescencia). La primera es la que afecta nuestro paciente.15
Recientemente, la relevancia clínica del nevus anemicus y el xantogranuloma juvenil, ha ido en aumento debido a su presencia en niños menores de 2 años, ya que, a estas edades los pacientes con NF de novo, por lo general cumplen un único criterio clínico, lo que resulta en retrasos diagnósticos. Ferrari y col proponen añadir nevus anemicus y xantogranuloma como criterios menores, debido a que tienen una asociación demostrada con NF1 y se han notificado en pacientes jóvenes. 16,17
En resumen, la enfermedad debería ser sospechada en cualquier paciente, que se presente con un tumor dérmico bien delimitado o un glioma del SNC, con múltiples máculas café con leche y/o historia familiar de neoplasias neurales periféricas o centrales. Se debe examinar la piel detalladamente en busca de efélides axilares, inguinales y neurofibromas; también buscar nódulos de Lisch con una lámpara de hendidura y un examen físico y neurológico completos.
El tratamiento de la NF1 históricamente ha sido empírico, siendo la cirugía la primera opción para aquellos neurofibromas que comprometan funcionalidad, sean molestos desde el punto de vista estético o aumenten su tamaño, sugiriendo un cambio maligno. El abordaje debe ser multidisciplinario: consejo genético, dermatología, oftalmología, traumatología y ortopedia, neurología y oncología. En el tratamiento de las máculas “café con leche” es posible ofrecer láser, ya sea rubí Q, de colorante o Nd: YAG con resultados variables. 18 Los neurofibromas profundos se deben resecar cuando comprometen una estructura vital o el sistema nervioso (central o periférico). Asimismo, se debe ofrecer terapia psicológica con los padres.
COMENTARIO
El presente caso expone la expresividad variable, de las manifestaciones cutáneas y deformidades óseas atípicas expresadas en la unión de cifosis y escoliosis, poco característica de la NF1 y confirmando el riesgo de recurrencia, con transmisión vertical de la herencia autosómica dominante, recalcando que los antecedentes familiares son cruciales en el diagnóstico final.
En conclusión, la NF1 es una enfermedad multisistémica que requiere seguimiento multidisciplinario, pero en cuyo diagnóstico y seguimiento los dermatólogos tenemos un papel destacado.
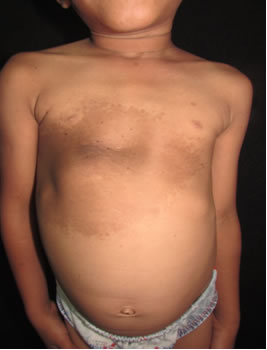
Fig 1: tórax anterior: se evidencia mácula que abarca desde línea axilar posterior derecha hasta la línea media clavicular izquierda, de 30 x 40 cm de longitud dentro de la que presenta área pardo oscura con pelos en su superficie desde su nacimiento, con aparición de pelos de un año de evolución.
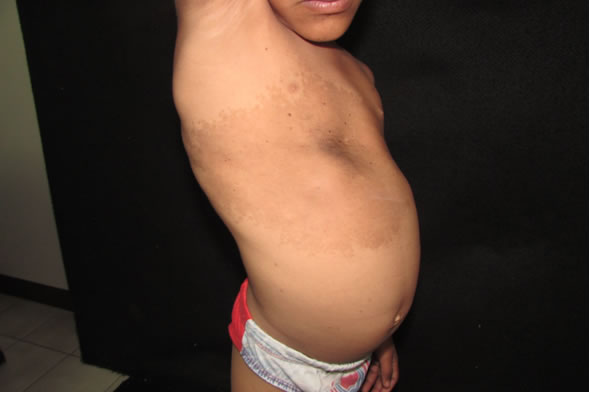
Fig 2: tórax anterior: mácula que abarca desde línea axilar posterior derecha hasta la línea media clavicular izquierda, de 30 x 40 cm de longitud.
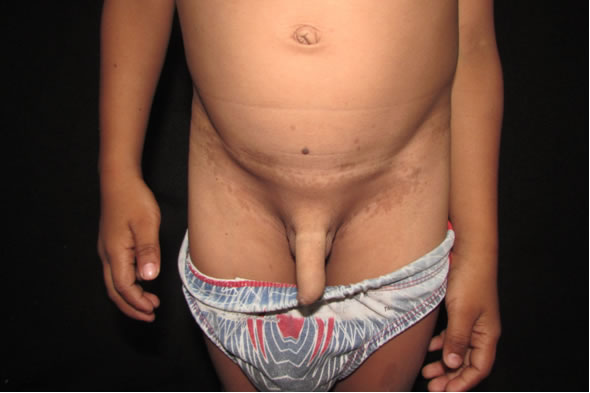
Fig 3: región inguinal con mácula café con leche, irregular, bien definida desde su nacimiento con efelides satélites.
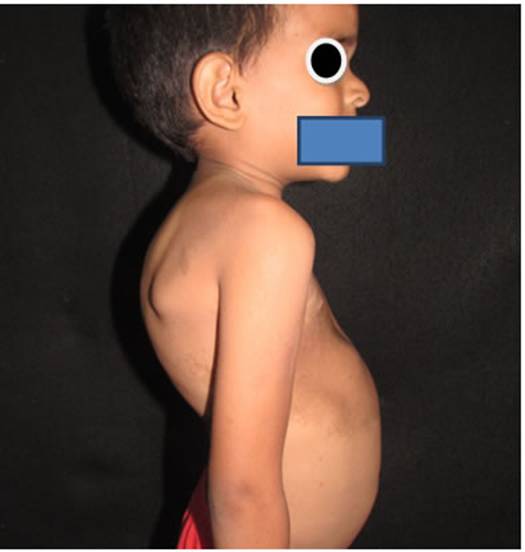
Fig 4: deformidad ósea caracterizada por aumento del diámetro ántero posterior del cráneo, desviación de la cintura escapular derecha cifoescoliosis y baja estatura proporcionada.
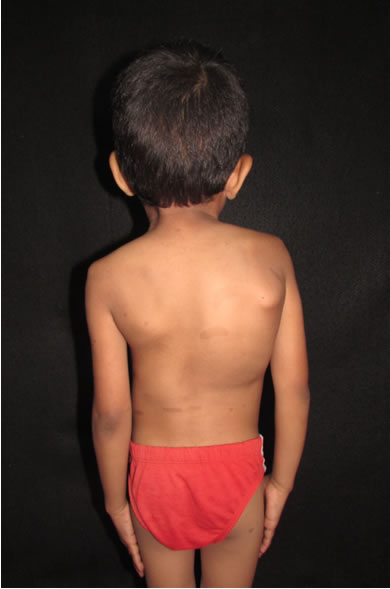
Fig 5: deformidad ósea caracterizada por desviación de la cintura escapular derecha cifoescoliosis, vista posterior.
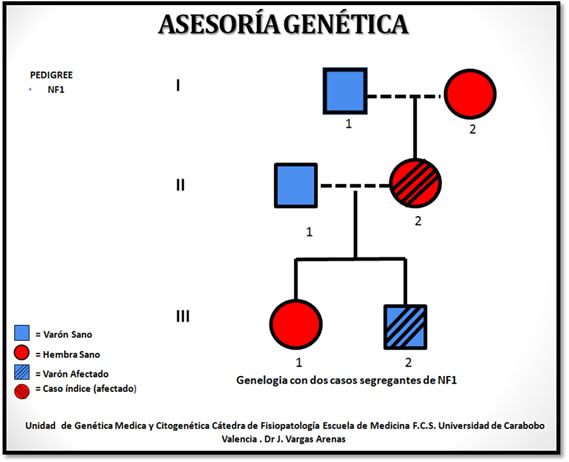
Gráfico I: Genealogía con dos casos segregantes de NF1, Unidad de Genética Médica y Citogenética Cátedra de Fisiopatología Escuela de Medicina F.C.S. Universidad de Carabobo Valencia. Dr J. Vargas Arenas.

