











 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Fabry pertenece al grupo de las enfermedades lisosomales, de patologías poco frecuentes de origen genético, debidas a mutaciones patogénicas en genes que codifican para proteínas, asociadas a la función de los lisosomas.1
Se debe a un error congénito del metabolismo glucofosfolipídico, consecuencia de un defecto en la actividad de la enzima lisosómica alfa-galactosidasa A; este déficit enzimático conduce al depósito progresivo de glucoesfingolípidos en múltiples tejidos, produciendo daño multisistémico progresivo. 2,3
A continuación presentamos el caso de un paciente de 43 años de edad, derivado a nuestra institución para el estudio de su enfermedad renal crónica. Tras el interrogatorio y un examen físico exhaustivo, se reúnen criterios que orientan al diagnóstico de enfermedad de Fabry.
CASO CLÍNICO
Paciente sexo masculino de 43 años de edad, oriundo de la Rioja, con antecedentes patológicos de hipertensión arterial (HTA) y accidente cerebro vascular isquémico de tres años previo a la consulta, con paresia braquio-crural derecha secuelar. Presenta cuadro de artralgias de cinco años de evolución, a predominio de las articulaciones interfalángicas de manos y pies, las que se exacerban ante cambios de temperatura.
Además presenta deterioro progresivo de la función renal, sin estudios previos. Su medicación habitual era ácido acetil salicílico 100 mg/24 hs, carvedilol 12,5 mg/12 hs, atorvastatin 10 mg/24 hs. En sus antecedentes familiares: madre fallecida a los 50 años por ACV, hermano con HTA, actualmente dos tíos maternos en hemodiálisis por padecer enfermedad renal crónica.
Se realiza laboratorio completo, orina de 24 hs, monoaminoxidasa. Ingresa con proteinuria de 300 mg/DL de creatinina, creatinina 4,43 mg/dl, urea 0,79 mg/dl.
Se solicita interconsulta con servicio de Oftalmología que informa córnea verticilata, el fondo de ojo evidencia forma y trayecto de vasos conservados. Es valorado por servicio de Neurología, donde solicitan RMN cerebral que informa: lesión cerebelosa derecha evolucionada e imágenes supratentoriales en sustancia blanca, que impresionan como isquemia crónica.
Al examen físico se evidenció facie con edema periorbitario, arco supraciliar pronunciado, orejas con lóbulos prominentes, puente nasal y base alar ancha, prognatismo, labios gruesos, un dismorfismo facial descrito como “pseudoacromegalia”. (Fig 1)
En el resto del tegumento se observan pápulas queratósicas, de color rojo-violáceo en región infraumbilical inguinal, genital, cara interna anterior y lateral externa de muslos y zona sacra, las que clínicamente impresionaban como angioquetaromas. Ante el diagnóstico presuntivo de enfermedad de Fabry, se solicita interconsulta con el servicio de Dermatología. (Figs 2 y 3)
Se toma biopsia de piel y el estudio histopatológico informa: en dermis papilar capilares dilatados, ectásicos e hiperqueratosis compacta, compatible con angioqueratomas. (Figs 4 y 5)
Se solicita biopsia renal por parte del servicio de Nefrología informando: glomérulo-esclerosis difusa, glomérulo con vacuolizacion citoplasmática de podocitos y células mesangiales, atrofia tubular moderada-severa y nefritis-túbulo intersticial moderada-severa. (Figs 6 y 7)
Se realizó screening de enfermedad de Fabry con examen de gota seca de sangre, para evaluar actividad enzimática de alfa galactosidasa con un valor hallado de 0,3 para un valor de referencia mayor o igual a 0,4.
Con los criterios reunidos arribamos al diagnóstico de enfermedad de Fabry.
DISCUSIÓN
La enfermedad de Fabry es un trastorno de almacenamiento lisosomal, ligado al cromosoma X causado por una actividad deficiente de α-galactosidasa, lo que resulta en la acumulación de glicoesfingolípidos con residuo terminal de α-D-galactosilo, particularmente globotriaosilceramida (GL-3, Gb3, CTH) y globotriaosylsphingosine (Lyso-GL-3, lyso-Gb3). Estos lípidos se acumulan progresivamente en la circulación y en prácticamente todos los tipos celulares y órganos, dando como resultado el desarrollo de un trastorno multisistémico.1,4
Las manifestaciones clínicas de esta enfermedad comienzan en la niñez y se caracterizan por la aparición de acroparestesias, dolor gastrointestinal, hiperhidrosis/anhidrosis, micro albuminuria y angioqueratomas aislados.1
La afectación cutánea es común y es uno de los signos clave, que puede llevar a un médico a sospechar de la enfermedad. Las manifestaciones de la piel incluyen: angioqueratomas, telangiectasias, sudoración anormal y linfedema. Estudios recientes de afectación cutánea en la enfermedad de Fabry, han demostrado que el espectro clínico de los angioqueratomas también es variado, suelen ser el primer signo de la enfermedad de Fabry y son secundarios a la acumulación de glucoesfingolípidos en la pared de los vasos sanguíneos, con posterior ectasia e hiperqueratosis.5
Las inclusiones de glucoesfingolípidos a nivel cutáneo, es posible objetivarlas con mayor detalle, utilizando la técnica de microscopía electrónica tanto en zonas comprometidas con angioqueratomas como en piel sana. A nivel endotelial se evidencian inclusiones vacuolares intracitoplasmáticas, electrodensas, en “cuerpos tipo cebra” con bandas alternas claras y oscuras.
El término angioqueratoma corporis difusum, puede sugerir que los angioqueratomas en esta enfermedad son múltiples, aunque no siempre es así; las lesiones pueden aparecer aisladas o agrupadas y pueden asentar en cualquier zona de la piel, las mucosas o genitales. Las descripciones clínicas clásicas destacan su distribución “en bañador “, ya que, con mayor frecuencia asientan en la zona de glúteos, los muslos y los genitales .4
En el estudio realizado por RM Guinovart, en la que incluía cinco pacientes con diagnóstico de angioqueratoma, demuestra que la edad promedio del inicio fue de 17,2 años con predominio en el sexo femenino. 5
Existen otras enfermedades de depósito lisosomal, que también presentan angioqueratomas generalizados como: fucosidosis, sialidosis, aspartilglucosaminuria y gangliosidosis GM1. Por ello, deberá realizarse un adecuado diagnóstico diferencial.6
Otras manifestaciones que podemos encontrar en la EF, son las acroparestesias que ocurren en la mayoría de los pacientes y se caracterizan por dolor urente insoportable, que puede ser continuo o episódico. El dolor afecta típicamente primero a los pies, seguido por las manos y puede desencadenarse por el ejercicio, el estrés y las temperaturas ambientales extremas, tal como el caso de nuestro paciente.3
Los hallazgos oftalmológicos consisten en tortuosidad de los vasos retinianos y conjuntivales y córnea verticilada (opacidad corneal caracterizada por líneas que se irradian desde el centro de la córnea); se manifiesta en el 77% de las mujeres y en el 73% de los hombres con enfermedad de Fabry2La pérdida de audición progresiva también es frecuente.
Si no se diagnostica y se trata adecuadamente la enfermedad, puede evolucionar a daño cardíaco y renal progresivo. La afectación renal se manifiesta con proteinuria progresiva, disminución del filtrado glomerular y nefropatía terminal entre la segunda y la cuarta décadas de la vida.
A nivel cardiovascular es frecuente que estos pacientes presenten hipertensión arterial, miocardiopatía hipertrófica progresiva, defectos de conducción, arritmias y valvulopatías (insuficiencia o estenosis).
Los pacientes son propensos a sufrir accidentes cerebrovasculares isquémicos, sobre todo en la cuarta y quinta décadas de la vida. Los pacientes afectados presentan un riesgo estimado de sufrir un accidente cerebrovascular isquémico y de ataques isquémicos transitorios, veinte veces superior respecto de la población general. En la resonancia magnética a menudo se observan lesiones cerebrales asintomáticas, típicamente en la sustancia blanca.
Después de la evaluación clínica, la determinación de la actividad de alfa-galactosidasa A, es el primer paso en el diagnóstico de laboratorio. Actualmente, la medición de la actividad de la enzima alfa-galactosidasa en leucocitos o cultivo de fibroblastos cutáneos en una gota de sangre seca, representa el gold standard del diagnóstico 2, mientras que la confirmación definitiva se obtiene al demostrar mutaciones patogénicas en los genes, que codifican las enzimas deficitarias.3
En cuanto al tratamiento, el reemplazo enzimático con infusiones intravenosas de alfa-galactosidasa A humana recombinante, disminuye los niveles de Gb3 en plasma y puede revertir el almacenamiento del sustrato en los lisosomas de las células endoteliales vasculares, por lo que con estas medidas es posible mejorar sustancialmente la calidad y esperanza de vida de los pacientes.1 También se ha recomendado terapia de reemplazo enzimático con agalsidasa alfa o beta, a dosis de 1 mg/kg de peso cada 15 días en infusión intravenosa 1,7, habiéndose con esta técnica demostrado que se estabiliza la función renal y se reduce la hipertrofia del ventrículo izquierdo; sin embargo, ninguna de las dermatosis desarrolladas en pacientes con EF previas al tratamiento sustitutivo, fue modificado por éste. 7
El tratamiento convencional del resto de las manifestaciones, requiere la administración de antiagregantes plaquetarios, para prevenir los accidentes cerebrovasculares o inhibidores de la enzima convertidora de angiotensina y antagonistas del receptor de angiotensina, para tratar la hipertensión y conservar la función renal. El trasplante renal resulta imprescindible en los pacientes con nefropatía terminal.7
CONCLUSIÓN
Arribamos al diagnóstico de enfermedad de Fabry, ya que, en el caso de nuestro paciente cumplía con criterios diagnósticos tales como: angioqueratomas, proteinuria, insuficiencia renal progresiva, anomalías oftalmológicas, acroparestesias, acv isquémico.
Resulta relevante destacar el rol del dermatólogo en conocer esta entidad, para sospecharla y poder instaurar un tratamiento de manera oportuna y así prevenir las complicaciones de la misma. Tanto la valoración familiar como el consejo genético revisten importancia, al tratarse de una patología ligada al cromosoma X.
El diagnóstico clínico de los angioqueratomas es fundamental, debido a que debe distinguirse de otras lesiones, tales como: angiomas eruptivos, púrpuras, petequias y otros angioqueratomas que pueden presentarse de manera solitaria, sin otra afección sistémica.
Por lo tanto, además del examen físico con los hallazgos cutáneos que orientan a la patología, es importante la evaluación global del paciente debido a su afección multisistémica, que requiere atención multidisplinaria por parte del equipo de salud.
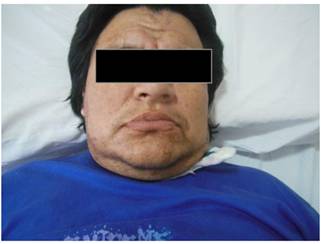
Fig 1:fascie descrita como pseudoacromegalia.

Fig 2 múltiples angioqueratomas en “traje de baño”.
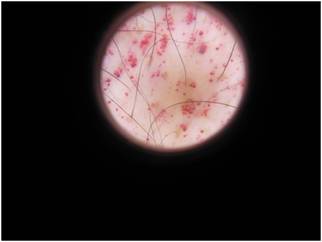
Fig 3:imagen dermatoscópica de angioqueratomas.
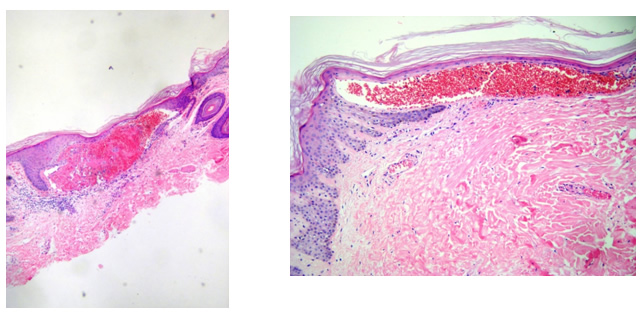
Figs 4 y 5:detalle anátomo-patológico de las lesiones cutáneas H/E 40X.
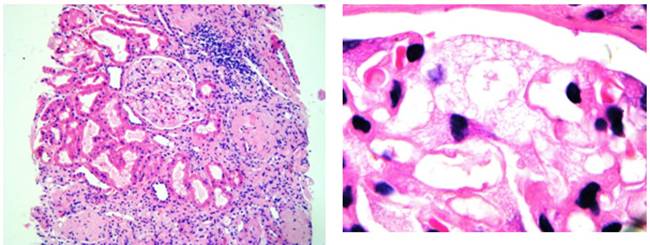
Figs 6 y 7:detalle anátomo-patológico del tejido renal H/E 40X.

