











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkCONCEPTO
Las porfirias son un raro grupo de trastornos metabólicos, provocados por deficiencias enzimáticas particulares de la vía de síntesis del grupo HEMO; así, se acumulan distintos sustratos moleculares que resultan en las manifestaciones clínico-morfológicas, propias de cada variedad de porfiria. Recientemente, se ha reportado la participación de otros genes que codifican proteínas, que no forman parte del mecanismo productor del HEM, pero se afirman causantes de formas concretas de la enfermedad. A la fecha, se han descrito cambios mutacionales que involucran a 7 genes: aminolevulinatodeshidratasa (ALAD), porfobilinógenodesaminasa (PBGD), uroporfirinógenosintetasa (UROS), uroporfirinógenodecarboxilasa (UROD), coproporfirinógeno oxidasa (CPOX), protoporfirinógeno oxidasa (PPOX) y ferroquelatasa (FECH), responsables de los tipos reconocidos de porfiria1.
La porfiriaeritropoyética congénita (PEC) o enfermedad de Günther, es un raro padecimiento heredado de manera autosómica recesiva dela dinámica metabólica de la porfirina; el defecto primario es la inactividad parcial de la enzima UROS III codificada en el cromosoma 10q25.3-q26.31. Aunque se manifiesta en la lactancia o la primera infancia puede diagnosticarse tardíamente; se caracteriza por graves episodios de fotosensibilidad cutánea cursando con formaciones ampollosas, edema y eritema intensos que resultan en mutilaciones cicatrizalesdesfigurantes, con pérdida relativa de los rasgos faciales; puede asociarse a zonas alopécicas, hipertricosis lanuginosa, afectación ocular con lesión corneal que puede llevar a la cegueray dentaria: tanto en la dentición primaria como secundaria con coloración café rojiza, que da coloración rojiza a la luz de Wood,por depósito tisular de uroporfirina I y coproporfirina. Otros hallazgos evidentes son: la anemia, esplenomegalia, porfirinuria y alteraciones esqueléticas como osteodistrofia y acroosteólisis2,3.
PRESENTACIÓN DE CASO
Se interna una escolar femenina de 11 años de edad, en el Servicio de Medicina Interna del Hospital,por presentar un grave cuadro de fotosensibilidad y mal estado general.En la interconsulta con Dermatología al examen físico, se observa dermatosis diseminada a zonas fotoexpuestas que involucran cara, cuello y extremidades superiores caracterizada por intenso edema tumefactodesfigurante y difuso, que compromete párpadoscon oclusión del eje visual asociado con una piel cetrino-eritematosa de aspecto poiquilodérmico, cursando con atrofia y cambios discrómicos moteados, además de formaciones erosivo-ulcerosas irregulares parcialmente cubiertas por costras hemáticas y necróticas, así como lesiones escaradas sobrepuestas y cicatrices varioliformes antiguas.Refiere sensación deardor de carácter urente; manifiesta fotofobia y queratoconjuntivitis arepetición (Fig 1). El resto de los hallazgos encontrados a la exploración fueron: deformidad y limitación anquilosante de los movimientos de las articulaciones metacarpo e interfalángicas de ambas manos, con discreto adelgazamiento esclerosante de la porción distal de los dedos (Fig 2).
Enel interrogatorio a los padres, no refierenninguna relación de consanguinidad, madre con diabetes mellitus tipo II e hipertensión arterial sistémica bajo control médico; de los antecedentes personales del paciente:leve retraso pondoestatural y psicomotor, así como anemia persistente de difícil control y episodios frecuentes de lesiones vésico-ampollosas que dejan una piel adelgazada, “acartonada”, manchada y concicatrices, tras la exposición lumínicosolar desde los 2 ó 3 años de vida. El padre refiere una coloración rojizo amarronada de la orina sin alguna sintomatología acompañante, tratada irregularmente como infección de vías urinarias.
Se solicitaron análisis generales de laboratorio, reactantes de fase aguda, exudado faríngeo, examen general de orina, pruebas de funcionamiento tiroideo, hepático y renal. Los resultados anormales significativos reportados son: Hb 8 g/dl, volumen corpuscular medio 70 fL, hemoglobulina corpuscular media 22 pg y reticulocitosis ++; el urianálisis con bilirrubinas (++/+++) y urobilinógeno de 16 mg/dl y uroporfirinas elevadas; resto de los estudios laboratoriales dentro de parámetros normales.
Con la finalidad de objetivar compromisode órganos internos, se solicitan además estudiosdeimagen, placa simple de tórax y ultrasonografía abdominal; sólo se observa cardiomegalia grado II a expensas de cavidades izquierdas y el ultrasonido sin alteraciones.
Los padres se negaron a la realización de biopsia de la piel involucrada, para análisis anátomo-patológico por considerarlo un procedimiento invasivo. En base a los datos clínicos muy sugerentes del padecimiento y su correlación bioquímica, se llegaal diagnóstico de porfiriaeritropoyética o enfermedad de Günther.
El tratamiento inicial en esta fase aguda consistió en: tres inyecciones vía intramuscular de fosfato sódico de dexametasona 5 ml por tres días consecutivos, además fomentos templados con sulfato de cobre al 1:1000 como antimicrobiano, astringente y antiinflamatorio tres veces al día durante 15 a 20 minutos cada una, con posterior aplicación de ácido fusídico y betametasona durante un par de semanas, con respuesta terapéutica favorable (Fig 3).
Como manejo de mantenimiento, se prescribe fotoprotectorde alta potencia en crema aplicado cada 2 horas en piel fotoexpuesta,además de medidas físicas de barrera como sombreros y paraguas, extremando en lo posible la exposicón a la luz solar, además de beta carotenos tabletas a razón de 50 mg al día. En las visitas periódicas de control a los Servicios de Dermatología y Medicina Interna, para evaluar el curso del padecimiento, la frecuencia e intensidad de los episodios inflamatorios, así como la existencia de hemólisis u otras manifestaciones sistémicas, se evidenció estabilidad de la enfermedad (Fig 4).
COMENTARIO
La porfiriaeritropoyética congénita, fue la primera variedad de este raro complejo de enfermedades metabólicas, descrita por Günther en 1911; de hecho la menos frecuente, sólo 200 a 300 casos reportados. Es ocasionada por deficiencia de la enzima uroporfirinógenosintetasa uro-gen III, en la que participan más de 22 mutaciones génicas. Las primeras manifestaciones ocurren a edades tempranas de la vida, como a la así llamada “orina roja” que mancha los pañales, además de episodios frecuentes de fotosensibilidad dolorosa extrema, que resulta en cicatrices y aspecto poiquilodérmicofibroescleroso de la piel;también se observa destrucción y retracción anquilosante óseo-cartilaginosa especialmente en manos. Otros datos típicos son: la eritrodoncia, afectación ocular y anemia hemolítica de diferente consideración, que bien puede requerir transfusiones o esplenectomía 4,5,6.
El objetivo del presente trabajo es identificar e integrar este tipo de enfermedades, de presentación poco habitual en la práctica médica diaria.Es importante considerar los valores laboratoriales elementales solicitados, con el objeto de efectuar la debida correlación clínico-patológica de la entidad, así como diferenciarla entre las enfermedades afines (Cuadro N° I).
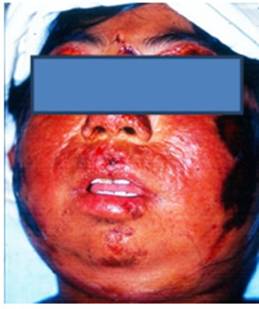
Fig 1: grave episodio deformante de fotosensibilidad.
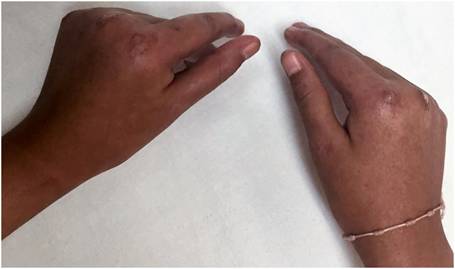
Fig 2: deformación y restricción del movimiento de las articulaciones.

Fig 3: aspecto clínico a los dos meses de tratamiento.

Fig 4: respuesta clínica al manejo con betacarotenos orales y estricta protección solar.
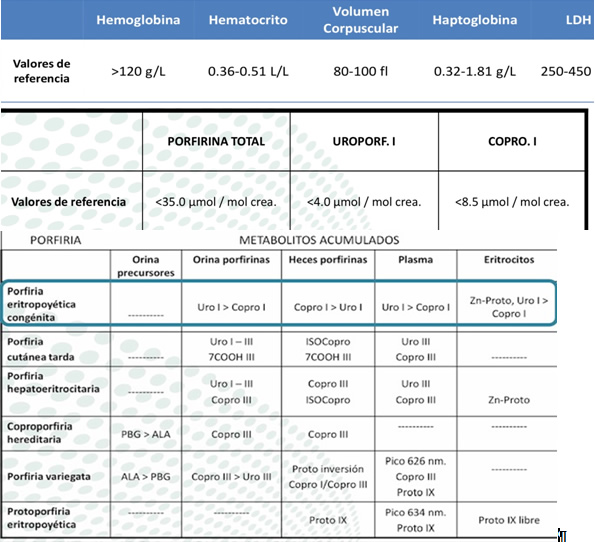
CUADRO N° I: Tipos de porfirias y valores de laboratorio

