Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El xantogranuloma juvenil es una entidad de etiología desconocida. Presenta una evolución benigna y autolimitada, con una involución espontánea en los primeros 5 años. Es una de las formas más comunes de histiocitosis de células no Langenhans.1
Si bien las lesiones pueden estar presentes desde el nacimiento, lo más común es que aparezcan durante el primer año de vida y en el sexo masculino.1,2 Las lesiones cutáneas pueden ser únicas o múltiples, de aspecto papulonodular, ovaladas, superficie lisa y consistencia duroelástica. Predominan en cuero cabelludo, cara, tronco y porción proximal de extremidades, y son asintomáticas.2,5
Si bien el compromiso extracutáneo es poco frecuente, hay que descartarlo cuando se presentan lesiones múltiples.1,2,5
El diagnóstico es clínico e histopatológico y son características las células tipo Touton.1,2,3,6,7 La inmunomarcación es positiva para CD68 y negativa para la proteína S-100 y CD1a, lo que la diferencia de las histiocitosis a células de Langenhans.3,4,5,7 Debido a su carácter autorresolutivo, no se dispone de un tratamiento específico. Puede realizarse tratamiento quirúrgico.1,2
CASO CLÍNICO
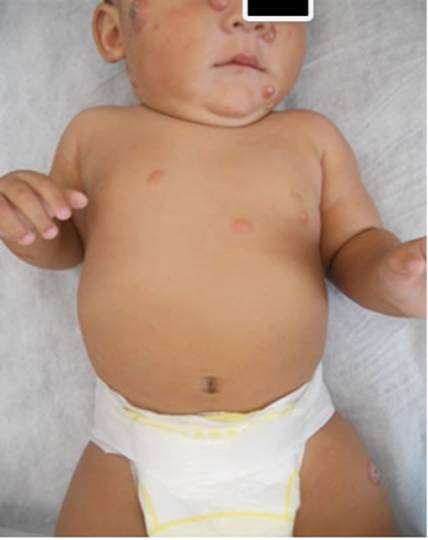
Paciente de sexo masculino, de 3 meses de edad, sin antecedentes perinatológicos ni personales de relevancia, consulta al servicio de dermatología por presentar múltiples lesiones tumorales, eritematosas, de consistencia duroelástica, asintomáticas, de aproximadamente 2 meses de evolución. Dichas lesiones se encontraban distribuidas en cuero cabelludo, cara y miembros. Se plantean como diagnósticos presuntivos xantogranuloma juvenil múltiple, histiocitosis de células de Langerhans y leucemia cutis. Se solicitan como estudios complementarios: valoración oftalmológica, que resultó normal; radiografía de cráneo, en la cual no se evidencian imágenes osteolíticas; laboratorio completo, en el cual se evidencia leucocitosis, por lo que se realiza interconsulta con hematología, con resultado de frotis normal y valores de leucocitosis en descenso en los controles posteriores. Se realiza biopsia de piel y en el informe se obtiene infiltrado de histiocitos, algunos de ellos vacuolados, y células gigantes multinucleadas de Touton en dermis. La población histiocitaria resultó positiva para CD 68 y negativa para el CD1a y la proteína S100, con lo que se confirmó el diagnóstico de xantogranuloma juvenil múltiple.
El paciente presenta aumento del número y tamaño de las lesiones en los sucesivos controles, por lo que se solicitan estudios complementarios para descartar compromiso sistémico. Se solicita nuevo control oftalmológico a los 6 meses, con resultado normal; ecografía abdominal, ecografía cerebral, tomografía axial computada de tórax y evaluación cardiológica. Todos los estudios dan resultados dentro de límites normales. Queda pendiente la realización de resonancia magnética nuclear de cerebro.
En la evolución se observan múltiples lesiones generalizadas; algunas de ellas presentan lesión costrosa central y en otras se evidencian pequeñas ulceraciones en su superficie. Se decide instaurar tratamiento con corticoides tópicos (betametasona- mometasona) en algunas de las lesiones. En la evolución puede observarse disminución de tamaño y aplanamiento de las lesiones tratadas.
DISCUSIÓN
El xantogranuloma juvenil es una enfermedad poco frecuente, de curso benigno y autolimitado que afecta a la piel y ocasionalmente, otros órganos.5 Predomina en lactantes de sexo masculino siendo la más frecuente de las histiocitosis de células no Langerhans.1
Puede estar presente desde el nacimiento, pero lo más común es que aparezca durante el primer año de vida, y que se presenten brotes durante meses y/ o años. En un pequeño porcentaje aparece en la edad adulta.1,2,8
Su etiología es desconocida, aunque presenta una histología característica constituida por proliferaciones reactivas benignas de elementos macrofágicos. Clínicamente se presentan únicas o, menos frecuentemente, múltiples neoformaciones de aspecto pápulo-nodular, rosadas o rojas en las etapas iniciales; posteriormente, amarillentas.2,4,5 Son de forma oval, superficie lisa, de 0.5 a 2 cm de diámetro, consistencia duro-elástica, indoloras al tacto y desplazables sobre planos profundos. Predominan en cuero cabelludo, cara, tronco y porción proximal de extremidades.2,5,8
Si bien el compromiso extracutáneo es poco frecuente, hay que descartar en pacientes con xantogranulomas múltiples; la córnea es el órgano más afectado, lo que puede llevar a hifema, glaucoma, hasta la pérdida de la visión.1,2,5,7,8. También pueden afectarse pulmón, hígado, sistema nervioso central, pericardio y huesos, y entonces se denomina xantogranuloma juvenil diseminado.2,6Habría una fuerte asociación entre el xantogranuloma juvenil múltiple y leucemia mielocítica crónica juvenil, sobre todo en pacientes con diagnóstico de neurofibromatosis de tipo 1.1,3,6,8
Para el diagnóstico, además de la clínica es de utilidad la histología, en la cual se visualizan en lesiones maduras, un infiltrado granulomatoso, células espumosas y células gigantes tipo cuerpo extraño y tipo Touton.1,2,3,6,7,8La inmunomarcación es positiva para CD68 y negativa para la proteína S-100 y CD1a, lo que la diferencia de las histiocitosis a células de Langenhans.3,4,5,6,7
Deben plantearse diagnósticos diferenciales con xantomas, mastocitomas, histiocitosis cefálica benigna, histiocitosis de células de Langerhans y dermatofibroma.1 El pronóstico es bueno: habitualmente las lesiones involucionan espontáneamente en 3-6 años después de su inicio. En algunos casos dejan atrofia cutánea, anetodermia y alteraciones en la pigmentación.1,2
No se dispone de un tratamiento específico; algunas lesiones tienen posibilidades de exéresis quirúrgica.1,2
CONCLUSIÓN
Se presenta una patología poco frecuente, generalmente benigna y autolimitada. En el caso de nuestro paciente, por múltiples lesiones generalizadas se solicitaron estudios complementarios para descartar compromiso sistémico, y debido a que presentó lesiones en aumento se decidió instaurar tratamiento tópico con corticoides de mediana potencia. Si bien no se puede saber si no correspondió a la involución espontánea de las lesiones, luego del tratamiento instaurado se observó aplanamiento y franca disminución de tamaño de las lesiones tratadas.