











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La epidermolisis bullosa (EB) comprende un grupo de enfermedades hereditarias raras con mutaciones genéticas que codifican proteínas estructurales que conducen a la fragilidad del epitelio con formación de ampollas y erosiones ante cualquier trauma físico. Aparece desde el nacimiento o la primera infancia, provoca pérdida de las propiedades de la piel y afecta también la córnea y las mucosas. El término "epidermólisis" describe el desprendimiento de epidermis y "bullosa", la formación de ampollas.1
Las estadísticas de la enfermedad son difíciles de estimar debido a problemas de reclutamiento y seguimiento de pacientes, las altas tasas de mortalidad y las diferentes posibilidades terapéuticas entre áreas geográficas. Estudios en Estados Unidos y en el Reino Unido estimaron una prevalencia de 11 casos por 1 millón de nacidos vivos (C1MRNV)2 y de 15 a 32 C1MRNV,3 respectivamente. En Colombia hay reportados sólo algunos casos en recién nacidos. 4, 5
Desde 2013 se describen 4 tipos de la enfermedad, que afectan a ambos sexos por igual, con mutaciones espontáneas, que son más comunes en la forma simple (EBS) pero raras en el tipo distrófico (EBD).6 La EBS es el tipo más común, en el 70% de los casos con lesiones en epidermis que no dejan cicatriz, y las complicaciones son raras; sus mutaciones en genes son EXPH5, KRT5 KRT14, TGM5. 1,7,8 La EBD es la menos común; en 5% de los pacientes, las lesiones están debajo de la membrana basal cutánea y dejan cicatrices que causan contracturas de manos, pies y otras articulaciones. La mutación genética es COL7A1. 1,7,9 La EBD también puede tener un patrón recesivo llamado Hallopeau-Siemens, que es la forma grave generalizada, al igual que el tipo Juntural (EBJ) y el síndrome de Kindler, compartiendo complicaciones como estenosis del tracto gastrointestinal, desnutrición, fallo de medro, anemia y carcinoma de células escamosas.7 La EBJ causa el 25% de los casos; las lesiones son en la lámina lúcida y generalmente deja cicatrices. Las mutaciones genéticas son LAMB3 (70% de los casos), COL17A1, LAMC 2 y 3. 1,7 El síndrome de Kindler tiene lesiones en diferente estado de evolución en y / o debajo de la membrana basal.7
Independientemente del tipo de enfermedad, ocurren importantes complicaciones: desnutrición, debida a limitaciones de alimentación por lesiones orales y estenosis esofágica, aumento del consumo de energía y pérdida de calor y nutrientes; estado hipercatabólico, causado por inflamación e infección de la piel; anemia crónica, debida al déficit nutricional o infeccioso y por pérdida de sangre durante las curaciones de las lesiones.10
La infección cutánea es frecuentemente causada por Staphylococcus aureus, meticilino sensible y resistente, Streptococcus pyogenes, Pseudomonas aeruginosa, Escherichia coli, Proteus mirabilis y Pseudomonas resistentes a la ciprofloxacina.10
Diagnóstico
El método de elección es el análisis genético mutacional en muestra de sangre. Debe hacerse en recién nacidos con sospecha clínica de la enfermedad y/o que sean parte de familias con antecedente de un niño con una enfermedad grave o con EB.11 Para la clasificación se utiliza la detección de antígeno por inmunofluorescencia y/o visualización directa, por microscopio electrónico de transmisión, de una muestra tomada de las ampollas.7 Para el diagnóstico prenatal se utilizan piel fetal o vellosidades coriónicas.12
Tratamiento
No se ha determinado una terapia curativa y el único tratamiento sería la terapia génica, que aún está en investigación. Otras terapias en estudio son el trasplante de células madre o de médula ósea, la terapia de reemplazo con proteínas recombinantes y el tratamiento con antiinflamatorios y antifibrinolíticos. 1,13
En cuanto al cuidado de la piel, es esencial la prevención de nuevas lesiones, minimizando la presión y el trauma. Cuando hay lesiones, el objetivo es reducir el dolor, prevenir y tratar complicaciones y mantener un buen estado nutricional. El manejo debe realizarse en un centro especializado con un equipo multidisciplinario que incluya nutrición, pediatría, dermatología, fisioterapia, enfermería, trabajo social, entre otros.1 ,4
El drenaje de las ampollas debe hacerse preservando el "techo de piel" que las cubre, para evitar su crecimiento y la aparición de nuevas. Luego se cubre con gasa no adhesiva; la frecuencia de cambio de los apósitos solo debe ser diaria si hay infección. Se debe evitar el desbridamiento del tejido necrótico, pero cuando sea necesario se debe hacer suavemente conjunto con emolientes; en raras ocasiones puede ser necesario aplicar sustitutos de la piel. 14,15 El baño debe hacerse con agua tibia y con emoliente a base de aceite, o si hay infección, con solución antiséptica como la clorhexidina. Se debe hidratar la piel con hidrogel o vaselina. 14,15
Las familias necesitan asesoramiento genético y entrenamiento en el cuidado de la piel, prevención y tratamiento de las complicaciones.
Presentación del caso
Se presenta el caso de dos pacientes recién nacidas, fruto de una gestación bicorial biamniótica, de una madre de 23 años con tamizaje prenatal infeccioso negativo, y con historia de un hijo previo del mismo padre, quien falleció con aplasia cutánea no confirmada a los 6 días de vida.
Las pacientes tuvieron buena adaptación neonata, peso y talla adecuados. Debido a las ampollas generalizadas fueron remitidas a nuestra institución para recibir atención especializada, con aproximadamente el 40% de área de superficie corporal (ASC) comprometida, con lesiones de mal olor y purulentas en tórax anterior y posterior, extremidades, genitales, cuero cabelludo, mucosa oral y nasal.
En forma secundaria a sus propios movimientos y secreciones corporales, aparecieron más lesiones junto con el crecimiento de las antiguas, lo que comprometió hasta un 80% de ASC. Las lesiones en mucosa nasal y oral empeoraron, con posterior desfacelación que causó inflamación y obstrucción local. (Figuras 1 y2)
Luego de revisar los posibles diagnósticos diferenciales como pénfigo vulgar, enfermedades autoinmunes crónicas, lupus eritematoso sistémico ampolloso y eczema dishidrótico vesicular, se concluyó una alta probabilidad de EB y se envió el estudio genético. Éste llegó positivo: Panel genético por NGS para Epidermolysis Bullosa heredada, variante previamente no informada, en el gen COL7A1 c.4738delG, p. V1580F para EBD, en homocigosis, una mutación patógena que produce una proteína truncada.
Las pacientes desarrollaron sepsis de origen cutáneo, con necesidad de antibióticos; pérdida de peso, con requerimiento de nutrición parenteral; y anemia, con necesidad de transfusiones de glóbulos rojos. En el manejo se hizo énfasis en el buen control del dolor.
Una de las pacientes desarrolló neumonía bacteriana con progresión a falla respiratoria y finalmente la llevó a la muerte a los 36 días de edad. La paciente que sobrevivió tenía un estado nutricional adecuado a los 8 meses de edad, aun presentando nuevas lesiones y cicatrices, pero sin infección o complicaciones graves.
Discusión
La EB es causada por mutaciones en genes que codifican proteínas estructurales, las que conducen a la fragilidad de los tejidos epiteliales y separación de las capas de la piel con la formación de ampollas, descamación y erosiones ante cualquier trauma físico. La EB compromete piel, córnea, mucosa gastrointestinal, genitourinaria y respiratoria. La gravedad del compromiso dependerá del tipo de enfermedad: EBS es la más común y la EBD, la menos común. 11,12 Puede haber complicaciones graves que pueden llevar a la muerte. 1,7,10
Se presenta el caso de dos recién nacidas gemelas con confirmación genética para EBD, con una variante previamente poco conocida llamada variante Hallopeau-Siemens. 7,9 Clínicamente se caracteriza por ampollas generalizadas desde el nacimiento, que progresan en número y extensión, y causan cicatrices, ulceraciones, sindactilia post-cicatrización, y deformidades y contracturas en flexión. Puede haber defectos en el esmalte dental; estenosis y cicatrización en córnea, faringe, esófago, uretra y ano; desnutrición, falla de crecimiento, anemia y carcinoma de células escamosas.7
Como aún no existe una terapia curativa, el tratamiento se basa en el adecuado manejo de las lesiones para prevenir infecciones y complicaciones, sumado a una buena analgesia y soporte nutricional, para reducir la morbimortalidad. Es muy importante proporcionar información y capacitación a las familias. 1,13,4
Conclusión
En el mundo se desconoce la incidencia y prevalencia real de la EB. La publicación de estos casos ayuda al personal que forma parte del cuidado de estos pacientes a obtener un diagnóstico oportuno, a conocer la enfermedad y a su tratamiento. Por ahora, el manejo se basa en prevenir y tratar complicaciones, disminuir el dolor y entrenar a las familias.
- Los padres dieron su consentimiento para datos, imágenes y revisión de historias clínicas.
FIGURAS
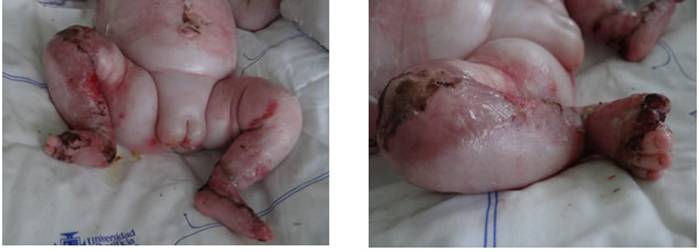
La gemela #1 presentó un compromiso más extenso y grave. Tenía lesiones en todas las
extremidades, incluidos los dedos y uñas, en diferentes estados de evolución; algunas, ya en resolución con cicatrices y otras, con tejido de granulación (1A). Las lesiones del miembro inferior derecho tenían tejido de granulación y edema, con desfacelación hasta el tercio proximal del muslo, con tejido de re-epitelización, y el hallux con costra seca (1B).

La gemela #2 Presentó un compromiso generalizado, pero con mayor área de piel sana y lesiones en etapas menos avanzadas que su hermana. Se muestran áreas de desfacelación en tórax con tejido de granulación. En el dorso de ambas manos, áreas de desfacelacion rodeadas de zonas en resolución y cicatrización. En la región distal de los antebrazos, lesiones con costra mielicérica (2A). Se evidencia lesión extensa en el miembro inferior izquierdo, con múltiples ampollas en el muslo y en la parte distal de la extremidad. Se observa tejido de granulación a nivel del maléolo izquierdo con área de contractura (limitación para el movimiento de extensión). Hay lesiones en los dedos de los pies que respetan el área plantar, con tejido necrótico (2B).
REFERENCIAS
1.Servicio Andaluz de Salud. Guía de práctica clínica para el cuidado de la piel y mucosas en personas con epidermolisis bullosa. Andalucía, España. 2009. Ed. Servicio Andaluz de Salud. Consejería de Salud. Junta de Andalucía
2.Fine J-D. Epidemiology of Inherited Epidermolysis Bullosa Based on Incidence and Prevalence Estimates From the National Epidermolysis Bullosa Registry. JAMA Dermatology. 2016; 152(11): 1231.
3.Browne F, Heagerty AH, Martinez A et al. The epidemiology of epidermolysis bullosa in the U.K.: A 9-year study. Br J Dermatol. 2011; 165.
4.Galán Gutiérrez S, Cesar J, Suárez M, Noval T et al. Epidermólisis bullosa de Herlitz en el paciente pediátrico: implicaciones anestésicas. Rev Colomb Anestesiol. 2014; 42(2): 132-135. Disponible en http://www.redalyc.org/pdf/1951/195131202012.pdf.
5.Millán MT, Millán MCT, Arias CC, González ML. Epidermólisis bullosa en un recién nacido. Reporte de un caso. (Epidermolysis bullosa in a newborn, a case report). CES Med. 2011; 25(2): 221-230.
6. Fine J-D. Open Access REVIEW Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010; 5. Disponible en http://www.ojrd.com/content/5/1/12.
7.Fine J-D, Bruckner-Tuderman L, Eady RAJ, Bauer EA et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification- ClinicalKey. J Am Acad Dermatol. 2014; 70: 1103-1126.
8.Pfendner EG y Bruckner AL. Epidermolysis Bullosa Simplex. University of Washington, Seattle. EE.UU., 1993. Disponible en http://www.ncbi.nlm.nih.gov/pubmed/20301543.
9. Pfendner EG y Lucky AW. Dystrophic Epidermolysis Bullosa. University of Washington, Seattle; 1993. Disponible en http://www.ncbi.nlm.nih.gov/pubmed/20301481.
10. Fine J-D y Mellerio J. Extracutaneous manifestations and complications of inherited epidermolysis bullosa- ClinicalKey. J Am Acad Dermatol. 2009; 61(3): 387-404. Disponible en https://www.clinicalkey.es/#!/content/playContent/1-s2.0-S0190962209007002?returnurl=null&referrer=null.
11. Has C. Molecular genetic assays for inherited epidermolysis bullosa. Clin Dermatol. 2011; 29(4): 420-426.
12.Fassihi H y McGrath JA. Prenatal Diagnosis of Epidermolysis Bullosa. Dermatol Clin. 2010; 28(2): 231-237.
13.Uitto J, Tuderman LB, Christiano AM et al. Progress Towards Treatment and Cure of Epidermolysis Bullosa: Summary of the DEBRA International Research Symposium EB2015 HHS Public Access. J Invest Dermatol. 2016; 136(2): 352-358.
14. Lara-Corrales I, Arbuckle A, Zarinehbaf S y Pope E. Principles of Wound Care in Patients with Epidermolysis Bullosa. Pediatr Dermatol. 2010; 27(3): 229-237.

