











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las enfermedades ampollares autoinmunes (EAA) comprenden un conjunto heterogéneo de patologías con baja frecuencia de presentación, que se caracterizan fundamentalmente por la presencia de ampollas, vesículas y/u otras lesiones en las superficies mucosas y/o cutáneas, y que se producen como consecuencia de la presencia de anticuerpos contra antígenos específicos de proteínas localizados a nivel epidérmico o en la unión dermoepidermica.1,2La demostración por técnica de Inmunofluorescencia directa (IFD) o indirecta (IFI) de estos anticuerpos, resulta por lo tanto fundamental para el diagnóstico de las mismas.
Se trata de patologías de evolución crónica, con remisiones y exacerbaciones, que presentan importante morbimortalidad aun con tratamiento temprano y adecuado.2,3
Pueden clasificarse, en términos prácticos, en el grupo de los pénfigos y en el grupo de enfermedades ampollares subepidermicas3-6 de acuerdo con las características clínicas e histopatológicas. El primer grupo incluye al Pénfigo vulgar, Pénfigo foliáceo, Pénfigo por IgA y Pénfigo paraneoplásico, caracterizadas por la presencia de anticuerpos contra proteínas específicas de los desmosomas.5,7,8Por otra parte, las enfermedades ampollares subepidermicas comprenden a patologías que se producen por el efecto de determinados anticuerpos contra proteínas localizadas en la unión dermoepidermica. Entre ellas se encuentran el penfigoide ampollar, penfigoide cicatrizal o de las membranas mucosas, penfigoide gestacional, epidermólisis ampollar adquirida, enfermedad por depósito linear de IgA y dermatitis herpetiforme, para nombrar algunas de ellas.4,9-13
La frecuencia de estas patologías es variable de acuerdo con la población estudiada1,2y a la patología en particular. Pocos estudios se han realizado sobre la prevalencia de todo el espectro de las enfermedades ampollares a nivel mundial. Daneshpazhooh1 en 2012 y Sobhan2 y col en 2016, estudiaron estas enfermedades en la población iraní, mientras que en 2017 Mittal14y col lo hicieron en la India y Patsasi15 y col en Grecia. En Argentina Rosti16 y col en 2007 realizaron un estudio retrospectivo de 6 años con la caracterización de las enfermedades ampollares en una población hospitalaria, arrojando los primeros datos para la caracterización de estas patologías en nuestro medio. Ante la falta de mayores datos sobre este grupo de entidades en nuestro país es que surge la necesidad de este estudio, que nos permitiría una mayor caracterización de los pacientes con enfermedades ampollares autoinmunes y en un futuro sentar las bases para una mejor aproximación diagnóstica y terapéutica.
Definición de conceptos generales:
Control de la actividad de la enfermedad: tiempo en el que dejan de formarse nuevas lesiones y las antiguas empiezan a sanar.
Remisión completa: ausencia de lesiones nuevas o establecidas mientras el paciente está recibiendo la mínima dosis de su terapia.
Recaída: Aparición de ≥3 nuevas lesiones en un mes, que no sanan espontáneamente en 1 semana, o bien, la extensión de lesiones ya establecidas en un paciente que logró el control de la enfermedad.
OBJETIVOS
Objetivos Generales
Describir las características clínicas, histopatológicas y de inmunofluorescencia de los pacientes con enfermedades ampollares autoinmunes que consultaron al Hospital Privado Universitario de Córdoba durante el período de enero 2008 a febrero 2018.
Objetivos Específicos
Determinar la frecuencia de las enfermedades ampollares autoinmunes en general y de cada una de ellas en particular, en el Hospital privado Universitario de Córdoba, en el periodo de tiempo estudiado.
Describir las comorbilidades presentes para cada una de las EAA de nuestra población y determinar la frecuencia de las mismas.
Describir tratamientos realizados, tiempo de control de la actividad de la enfermedad con el tratamiento, tiempo desde iniciado el tratamiento hasta la remisión completa
Determinar recaídas de las patologías estudiadas, evaluar si existe relación entre las mismas y algún factor reconocido (tratamiento elegido, abandono de tratamiento, enfermedad concomitante)
MATERIALES Y MÉTODOS
Para el desarrollo de este trabajo se siguieron los lineamientos de la Declaración de Helsinki Finlandia, de la Asociación Médica Mundial (1964); revisada y enmendada por la 64º Asamblea General de Fortaleza, Brasil 2013.
El trabajo cuenta con la aprobación del Comité de Ética del hospital (anexo 3).
Se trata de un estudio retrospectivo, descriptivo y observacional.
La población analizada fueron todos los pacientes, de cualquier edad y sexo, que asistieron al servicio de dermatología del Hospital Privado Universitario de Córdoba (HPUC) en el período comprendido entre enero de 2008 y enero de 2018.
La muestra incluyó a aquellos pacientes que tuvieran diagnóstico clínico, histopatológico y de IFD de enfermedad ampollar autoinmune.
Para la obtención de datos se analizó la información de la historia clínica electrónica (HCE), que constituye el único depósito de información hospitalaria desde el año 2008. Se incluyó para el análisis aquellos pacientes que presentaban como problema en la HCE la palabra Pénfigo, Penfigoide, Dermatosis lineal por IgA, Epidermólisis ampollar adquirida, dermatitis herpetiforme, lupus ampollar. También se hizo una búsqueda mediante la base de datos del servicio de patología de nuestro hospital de dichos diagnósticos, tanto en los informes de la histopatología como en los de IFD realizada sobre piel no lesional. Una vez identificados los pacientes, se procedió a revisar las historias clínicas correspondientes.
Se seleccionó para el estudio sólo a aquellos pacientes que presenten diagnóstico clínico por un dermatólogo, diagnóstico histopatológico y de IFD de alguna de las EAA, que hayan sido realizados o confirmados en el HPUC, con tratamiento y/o seguimiento de por al menos 1 año en el servicio de Dermatología del HPCU.
Se confeccionó una ficha para cada paciente incluido en este estudio (ver Anexo) la que se completó con datos de la HCE.
Criterios de inclusión:
Pacientes con diagnóstico clínico de EAA que tengan histopatología e inmufluorescencia directa compatible
Pacientes que hayan hecho tratamiento y seguimiento por al menos un año en el servicio de Dermatología del HPCU
Pacientes en los que se puedan completar los datos de la ficha destinada para el estudio
Criterios de exclusión
Pacientes con diagnóstico clínico de alguna EAA, pero sin diagnóstico histopatológico y/o sin estudio de IFD correspondiente.
Pacientes a los que no se les pudiera completar los datos de la ficha confeccionada para este trabajo
Pacientes con enfermedades ampollares no autoinmunes.
Análisis estadístico: Con los datos recogidos con la ficha de recolección se creó una base de datos de tipo Excel que fue utilizada para su posterior procesamiento estadístico. Se calcularon frecuencias y porcentajes para las variables cualitativas, como el sexo, diagnóstico clínico de la enfermedad, diagnóstico histopatológico y de inmunofluorescencia. Media, moda y desvío estándar para las variables cuantitativas tales como edad, tiempo de evolución de la enfermedad al momento del diagnóstico, recaída de la enfermedad en meses, entre otros. Los resultados se presentaron en forma de gráficos o tablas, según corresponda. Para el análisis de la relación entre las recaídas y algún evento particular, se aplicó el test de chi-cuadrado y el test exacto de Fisher fue utilizado en caso de ser necesario. Se utilizó un nivel de significancia del 0,05. La información fue analizada con el programa SPSS versión 20.0 o superior.
RESULTADOS
Características generales de la muestra
Durante el período de tiempo estudiado consultaron al servicio de dermatología del HPUC 203.882 pacientes. La muestra final estuvo conformada por un total de 54 pacientes diagnosticados con una EAA en el período de tiempo estudiado, con una tasa de frecuencia relativa de presentación para todas las enfermedades ampollares autoinmunes del 0,02% en los 10 años estudiados, con lo que se estima una media de presentación de 5 pacientes con diagnóstico de EAA por año en el servicio de dermatología de nuestro hospital.
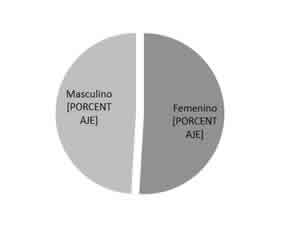
La edad media de los mismos fue de 62,13 años (D. E=21,7), con edades comprendidas entre los 21 y los 90 años. No tuvimos casos de niños en este estudio. Con respecto al sexo, la distribución fue muy similar, en el 50,9% fueron pacientes de sexo femenino y en el 49,1% restante de sexo masculino (Fig. 1).
El tiempo de seguimiento fue en promedio de 22,7 meses, con un mínimo de 12 meses y un máximo de 57.
El diagnóstico clínico inicial realizado por un dermatólogo del servicio fue correspondiente con el diagnóstico final en el 88,89%. En 6 casos 11,11% (cuatro casos de Penfigoide ampollar, interpretados inicialmente como farmacodermia y dos Dermatitis herpetiforme con diagnóstico presuntivo de eccemas) el estudio histopatológico y de inmunofluorescencia directa fue indispensable para arribar al diagnóstico definitivo.
Las características de los pacientes según diagnóstico se muestran en la tabla 1. No se encontraron casos diagnosticados como epidermólisis ampollar adquirida, pénfigo paraneoplásico ni penfigoide gestacional.
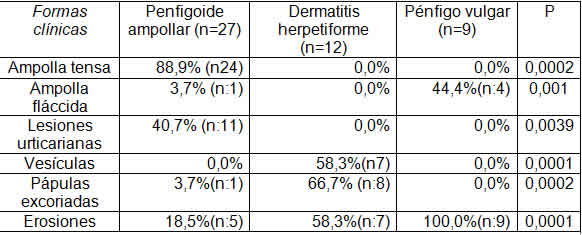
En cuanto a la forma de presentación clínica, en términos generales, casi la mitad de los casos presentaron erosiones (45,3%) que se observaron en todos los casos e incluso fueron las formas de presentación de los pacientes con Pénfigo foliáceo (1), Pénfigo por IgA (1), dermatosis lineal por IgA (2) y Lupus ampollar (1). Los pacientes con Penfigoide ampollar presentaron en su mayoría la forma clásica con ampollas tensas en el 88,9% y lesiones urticarianas en el 40,7%. La afectación mucosa ocurrió en el 78% de los casos de los pacientes con Pénfigo vulgar, tratándose de la mucosa oral en el 67% con un solo paciente que además presentó afectación de la mucosa genital (11%). No tuvimos casos de afectación de mucosa ocular y tres pacientes (un paciente con Pénfigo vulgar, un paciente con Dermatosis lineal por depósito de IgA y un paciente con Penfigoide de las mucosas) presentaron afectación de la mucosa genital. Los pacientes con dermatitis herpetiforme presentaron en su mayoría pápulas excoriadas y vesículas.
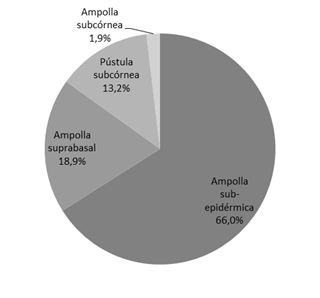
La mayoría de los pacientes que integraron este estudio, presentaron ampollas subepidérmicas, con el 66% de los casos (Fig. 2). Le siguieron en orden de frecuencia, las ampollas suprabasales (18,9%), las lesiones de tipo pústulas subcórnea (13,2%) y finalmente las ampollas subcórnea (1,9%). La tabla 2 muestra Las formas clínicas halladas para cada enfermedad.
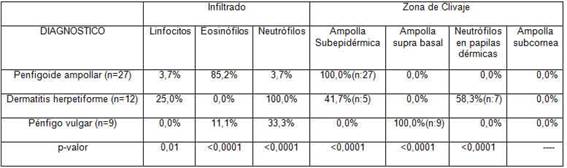
En la tabla 3 se muestran las características histopatológicas de los pacientes de nuestra muestra. El infiltrado inflamatorio predominante para los pacientes con penfigoide ampollar fue del 85,2% de eosinófilos con una minoría del 3,7% que presentó neutrófilos y linfocitos como infiltrado predominante. Los pacientes con dermatitis herpetiforme presentaron en un 100% de los casos infiltrado de neutrófilos, mientras que los pacientes con diagnóstico de pénfigo vulgar no presentaron infiltrado predominante en la mayoría de los casos, todos estos datos fueron estadísticamente significativos. En los demás casos no se pudo calcular el porcentaje por el tamaño de la muestra.
Las características de la IFD se encuentran en la tabla 4. La presencia de IgG y C3 ocurrió de forma lineal en la membrana basal en el 48,1% de los casos con penfigoide ampollar, un solo paciente presentó solo expresión de C3 y 12,56% (n:3) presentaron solo IgG. El 96,3% de los casos con pénfigo vulgar presentaron IgG interqueratinocitos 48,1% presentaron tanto IgG como C3 con esta disposición. Los pacientes con dermatitis herpetiforme presentaron en un 100% IgA, un 8,3% además presentó expresión de C3.
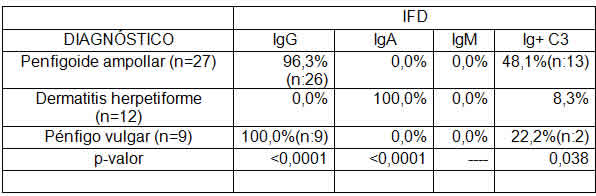
Tratamientos Realizados
En el 79,63% de los pacientes se utilizaron tratamientos sistémicos, la mayoría utilizó corticoides y azatioprina (Tabla 5). Los tratamientos tópicos alcanzaron al 69,8% de los pacientes, pero solo fueron exclusivos en el 20,37% de los casos en cinco pacientes con Penfigoide ampollar, un paciente con diagnóstico de Pénfigo vulgar y cinco pacientes con Dermatitis herpetiforme. El 55,81% de los pacientes utilizaron ambos tipos de tratamientos. Es importante destacar que todos los pacientes con diagnóstico de dermatitis herpetiforme se sometieron a una dieta libre de gluten, indistintamente del tratamiento tópico o sistémico indicado para su patología. No existieron casos de tratamientos con agentes biológicos y solo 3 pacientes (11,11%) con diagnóstico de Penfigoide ampollar tuvieron como tratamiento sistémico antibióticos del grupo de las tetraciclinas.
La duración media de la enfermedad, calculada como el periodo de tiempo comprendido entre el inicio de los síntomas y la remisión completa, fue de 13,04 meses (D. E=11,6) con valores comprendidos entre 1 y 55 meses de máximo.
En el 91% de los casos se llegó al control de la enfermedad, con una media de 2,9 meses. Sólo dos casos no alcanzaron el control a pesar del tratamiento y en tres casos este dato no fue establecido.
La remisión completa de la enfermedad se alcanzó en el 78,7% de los casos. El tiempo promedio de la remisión completa fue de 4,4 meses (D. E=5,1). (Tabla 6)
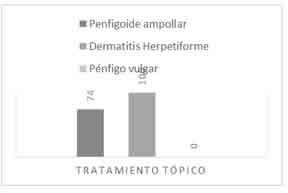
En el 38,3% de los pacientes hubo una recaída (Fig. 3), mientras que en la mayoría (61,7%) no la hubo.
Complicaciones
En relación con las complicaciones, un 14,95% de los pacientes tuvieron alguna complicación. Se detectaron infecciones en el 9,4% y el 1,85% (1 caso) presentó osteopenia, leucopenia, elevación de las enzimas hepáticas e insuficiencia renal respectivamente. Todos los pacientes que presentaron complicaciones las tuvieron durante el tratamiento en combinación con corticoides sistémicos y azatioprina. Hubo dos muertes debidas a septicemia, ambos pacientes recibieron tratamiento con corticoides y azatioprina, presentaron recaídas durante el tratamiento y tenían diagnóstico de Pénfigo vulgar.
Características particulares de los pacientes según diagnóstico:
De todas las EAA de nuestra muestra, el número de pacientes con Penfigoide ampollar, Dermatitis herpetiforme y Pénfigo vulgar fue significativamente mayor y nos permitió realizar un análisis estadístico más detallado que se describe a continuación.
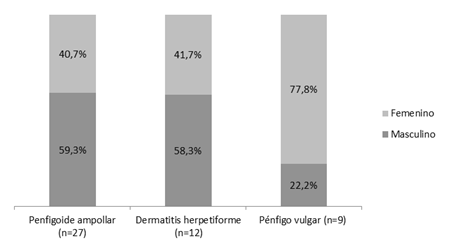
La frecuencia relativa de presentación en los 10 años fue del 0,01%, 0,006% y 0,004% para Penfigoide Ampollar, Dermatitis herpetiforme y Pénfigo vulgar, respectivamente tomando en cuenta el total de pacientes vistos en el servicio de dermatología del HPUC. Con respecto al total de EAA, el 50% fue diagnosticado como Penfigoide ampollar, el 22,2% como Dermatitis Herpetiforme y el 16,6% Pénfigo vulgar. Los resultados indicaron que, de los pacientes con Penfigoide ampollar y Dermatitis herpetiforme, la mayoría fueron de sexo masculino (59,3% y 58,3% respectivamente) mientras que en los pacientes con Pénfigo vulgar la mayoría fue de sexo femenino (77,8%). A pesar de las diferencias encontradas en esta distribución, al aplicar el test estadístico no resulto significativo el resultado (Fig. 4).
En cuanto a la edad de los pacientes por grupos, en los pacientes con penfigoide ampollar la media fue superior, de 78,4 años (DE=8,1). En el grupo con dermatitis herpetiforme, la edad media fue de 42,3 años (D. E=16,8) y para los pacientes con pénfigo de 49,2 años (D. E=22,3). Al aplicar el test estadístico correspondiente se concluye que las diferencias son significativas (p<0,0001).
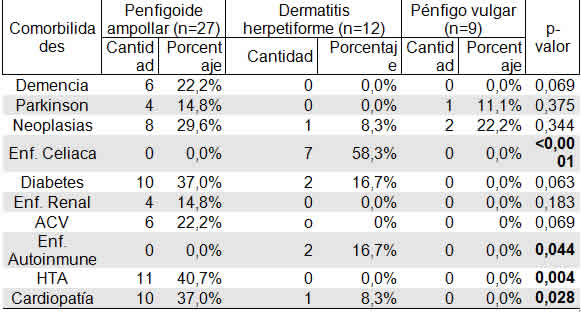
Al evaluar los porcentajes de comorbilidades en cada uno de los grupos diagnosticados (Tabla 7), que resultaron estadísticamente significativos, se pudo observar que en los pacientes con Dermatitis herpetiforme se presentó un alto porcentaje de enfermedad celíaca (58,3%) y de otras enfermedades autoinmunes (vitíligo y tiroiditis de Hashimoto en dos pacientes respectivamente) pero con un porcentaje menor (16,7%).
Casos de hipertensión arterial ocurrieron solo en los pacientes con Penfigoide ampollar (40,7%), y sin casos en los otros grupos. Así mismo, las cardiopatías, se dieron en un porcentaje superior en los pacientes con penfigoide ampollar (37%).
Los pacientes con Penfigoide ampollar y Pénfigo vulgar fueron los que más recibieron tratamiento sistémico, 85% y 100%, respectivamente (Fig. 5). Por otro lado, los tratamientos tópicos (Fig. 6) fueron superiores en la Dermatitis herpetiforme (100%) y en Penfigoide ampollar (74%). Ambos análisis resultaron estadísticamente significativos (p<0,05). Todos los pacientes con Dermatitis herpetiforme realizaron una dieta libre de gluten. El detalle de los tratamientos realizados en estas enfermedades se encuentra en la tabla 8.
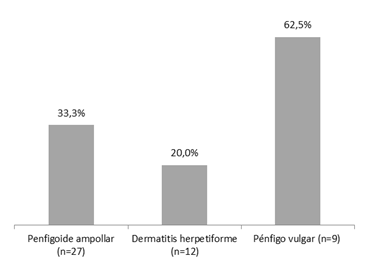
Se pudo observar que, de la totalidad de los pacientes diagnosticados con Penfigoide ampollar, hubo un 33,3% de recaídas, en la Dermatitis herpetiforme el 20% de estos pacientes presentó recaídas y, en los diagnosticados con Pénfigo vulgar, el porcentaje fue de 62,5% (Fig. 7). Las diferencias no resultaron estadísticamente significativas (p>0,05).
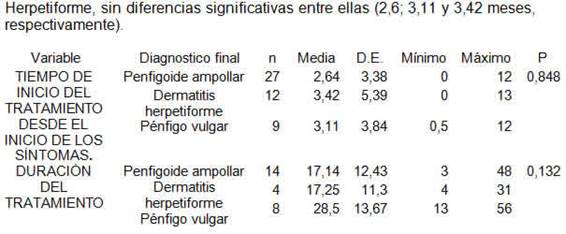
De los pacientes incluidos, 47 realizaron un seguimiento de la enfermedad hasta la remisión completa, 5 pacientes se perdieron en el seguimiento más allá del año. La duración media de la enfermedad fue en los pacientes con Penfigoide ampollar, fue de 10,5 meses (D. E=9,9), en la Dermatitis herpetiforme fue de 6,6 meses (D. E=4,8) y en el Pénfigo vulgar, 24,4 meses (D. E=15,1). Al aplicar el correspondiente test, las diferencias resultaron estadísticamente significativas (p<0,0001). En la Tabla 9 se encuentran datos del curso de las distintas EAA. Los pacientes con diagnóstico de Pénfigo vulgar presentaron una media de duración del tratamiento de 28.5 meses a diferencia de los pacientes con Penfigoide ampollar y Dermatitis Herpetiforme que presentaron una media de 17,14 y 17,25 meses respectivamente, valores que al análisis estadístico no presentaron significación (p:0.132). La media de tiempo desde el inicio de los síntomas al inicio del tratamiento fue similar en todos los casos de Penfigoide ampollar, Pénfigo vulgar y Dermatitis Herpetiforme, sin diferencias significativas entre ellas (2,6; 3,11 y 3,42 meses, respectivamente).
DISCUSIÓN
Las EAA son un complejo grupo de enfermedades cuya fisiopatogenia radica en la presencia de diversos anticuerpos contra componentes epidérmicos o de la unión dermoepidérmica. Representan un desafío en la práctica clínica por su elevada morbimortalidad.
La frecuencia de presentación y la incidencia de las EAA es muy variable de acuerdo con la población estudiada, ya que factores ambientales, étnicos y variaciones genéticas resultan importantes para el desarrollo de cada enfermedad2,19. Según lo publicado por Corach35 en 2010 sobre la composición genética de los argentinos, la población de la Argentina representa un mosaico complejo de contribuciones étnicas de orígenes diversos. Según este estudio la composición genética de los habitantes argentinos de la región centro de nuestro país, en la que se encuentra la ciudad de Córdoba, presenta una marcada herencia europea (80.73%) seguida por nativo-americana (16,21%) y afroamericana en un menor porcentaje (4,03%).
En nuestro estudio, la frecuencia de presentación de todas las EAA fue 0,02% en pacientes que consultan al servicio de dermatología del HPUC.
En este estudio, no existieron diferencias significativas con respecto al sexo, a
diferencia de lo que hallaron Daneshpazhooh1 y col y Sobhan2 y col en sus respectivos estudios retrospectivos sobre enfermedades ampollares autoinmunes en Irán con un predominio femenino en todos los grupos de EAA.
Para el diagnóstico y diferenciación de las EAA es imprescindible el estudio histopatológico y la demostración de anticuerpos tanto con técnica de inmunofluorescencia, directa e indirecta, o bien otras técnicas más complejas como electrotransferencia, Elisa, entre otras. En nuestro trabajo el diagnóstico clínico fue consistente con el diagnóstico histopatológico y de IFD en casi el 88,8% de los casos, similar a lo hallado por Sobhan2, basándose en la forma de presentación y en los síntomas acompañantes que se presentaron de forma similar en cada una de las entidades. Solo fue dificultosa la sospecha clínica de enfermedades que resultaron muy infrecuentes: dermatosis lineal por IgA y lupus ampollar (con solo un caso representativo de cada una), así como también de casos de penfigoide ampollar que fueron inicialmente diagnosticados clínicamente como farmacodermia símil penfigoide, pero que luego de un estudio histopatológico y de IFD fueron catalogados como penfigoide ampollar y de dos casos de Dermatitis Herpetiforme interpretados inicialmente como eccemas.
El periodo de tiempo entre el inicio de la clínica y el inicio del tratamiento fue de 3,05 meses, sin diferencia significativa entre las EAA. La duración de la enfermedad fue mayor para los pacientes con Pénfigo vulgar, similar a lo descripto por Sobhan2 y col quienes hallaron una media de 5,37 meses para el inicio del tratamiento en este grupo de pacientes, aunque luego del análisis estadístico, en nuestros casos, este valor no fue significativo.
Las recaídas se presentaron en todas las entidades, siendo mucho más frecuentes en los pacientes con Pénfigo vulgar (62,5%), seguido por el 33.3% de pacientes con Penfigoide ampollar y el 20% de los pacientes con Dermatitis herpetiforme. Estos datos condicen con los hallados en la literatura1,2que describen una tasa de recaídas del 75% para los pacientes con pénfigo vulgar, 57% para PA y 18% para Dermatitis Herpetiforme. No pudimos determinar asociación entre las recaídas y eventos conocidos (infección, abandono del tratamiento o comorbilidad exacerbada) por tratarse de una muestra de tamaño limitado y con pérdidas de datos en la recolección de estos.
Penfigoide ampollar
Se trata de la enfermedad más frecuente de nuestro estudio (n:27), lo que se corresponde con lo hallado por Bertram y col en su estudio sobre incidencia de EAA en Alemania3, lo descripto por Joly y col en Francia20 y lo hallado en un estudio del Reino Unido por Langan17 y col que evidenció una incidencia para el penfigoide ampollar de 4,3 por 100.000 personas año. Sin embargo, en países como Iran2, Kuwait33, India14 y Grecia15 prevalece el PV como entidad de mayor frecuencia, y el PA se ubica en el segundo lugar de frecuencia, debido probablemente a cuestiones étnicas y genéticas22.
La mayoría de nuestros pacientes con PA fueron hombres (59,3%), con una edad media de presentación de 78,4± 8,1 años, lo que se condice con lo encontrado por Bertam3 en Alemania y un estudio en nuestro país realizado por Coelho22 y col que valoraron las características clínicas del penfigoide ampollar en una población hospitalaria de Buenos Aires, Argentina, y encontraron una media de edad similar (75,97 años) pero con un predominio femenino (55,5%). En un estudio multicéntrico desarrollado en Francia20 la edad media de los pacientes era mayor (82,4) años y una amplia mayoría de los 201 casos pertenecían al sexo femenino.
Es sabido que la forma de presentación del PA inicia en la mayoría de los casos con lesiones urticarianas9,17,23,24que evoluciona a ampollas tensas, lo que en nuestro trabajo le sucedió al 52,8% de los casos, la presencia de ampollas tensas ocurrió en todos los casos de PA. Solo tuvimos un caso de presentación localizada y ningún caso con otras variantes como dishidrosiforme, vesicular o nodular entre otras. En la literatura consultada la gran mayoría de los pacientes presentan esta forma clásica en la clínica, aunque se describen mayor porcentaje de presentaciones localizadas (11,1%), vesicular (2,2%) y nodular 2,17.
La histopatología de los pacientes de PA presentó en todos los casos una ampolla subepidérmica. El infiltrado inflamatorio fue predominante en el 92,6% de los casos y se trató de infiltrado eosinofílico en el 85,2%. El patrón de IFD fue similar en todos los casos, con depósitos lineales de autoanticuerpos a lo largo de la zona de la membrana basal, presentando en todos los casos anticuerpos IgG y en el 48,1% también complemento C3. Estos resultados se condicen con los hallados en la bibliografía24,25.
Múltiples estudios se han realizado con el fin de evaluar la asociación del PA y otras comorbilidades21,23,24,26,27. Taghipour23 en su estudio de casos y controles sobre la asociación de PA, enfermedad cerebrovascular y demencia, encontró una asociación estadísticamente significativa entre estas dos enfermedades neurológicas y el desarrollo del PA. Bastuji-Gorin25 y col, al igual que Chen y col26 y Lloyd-Lavery y col 27 demostraron también la asociación con enfermedad de Parkinson en este grupo de pacientes. En nuestro trabajo los pacientes con PA presentaron enfermedad de Parkinson y demencia, pero sin asociación estadísticamente significativa, mientras que un alto porcentaje presentaron hipertensión arterial y cardiopatía, datos que concuerdan con la literatura24-27 pero que no se ha demostrado que tengan repercusión en la evolución de la enfermedad.
Con respecto al tratamiento utilizado, en nuestro trabajo el 81.5% realizó tratamiento sistémico, el 77,8% recibió conjuntamente corticoides tópicos y solo 3 pacientes utilizaron solo corticoides tópicos como terapéutica. Tres pacientes a su vez recibieron tratamiento con tetraciclinas, además del tratamiento tópico, hecho que se describe en diferentes estudios9, pero que hasta el momento no estaría indicado como primera línea. La media de duración del tratamiento de los pacientes fue 17,14 meses, un tiempo mucho mayor al encontrado por Sobhan2 que fue de 5.4 meses; otros estudios no presentan este dato.
Dermatitis Herpetiforme
La segunda entidad más frecuente en nuestro estudio fue la DH, con una frecuencia estimada del 22% de todos los casos de EAA. La mayoría de los casos (53,8%) fueron del sexo masculino, lo que difiere a lo hallado en la literatura.1,2 La media de edad fue de 42,3 años, superior a la descripta en el estudio de Fuertes29 y col en el hospital Clínic de Barcelona, en el que la media de edad fue de 30 años. El 58,3% de nuestros pacientes tenían asociada enfermedad celíaca (5 pacientes presentaron el diagnóstico previo a la sintomatología cutánea y 2 pacientes fueron estudiados y diagnosticados una vez hecho el diagnóstico de DH), datos que se corresponden con la literatura que sitúa esta asociación entre el 60 y 75% de los casos31,32 un porcentaje menor (16,7%) pero estadísticamente significativo, presentó enfermedad autoinmune al momento del diagnóstico. Fuertes29 y col también encontraron estas asociaciones, aunque con mayor frecuencia que en nuestro estudio (82% de pacientes con enfermedad celíaca y 39% con enfermedad autoinmune asociada). No observamos otras comorbilidades asociadas en los pacientes con este diagnóstico.
La mayoría de nuestros pacientes presentaron pápulas excoriadas y erosiones como forma clínica, solo 2 casos presentaron vesículas, presentando además la mayoría prurito como signo acompañante.
En la histopatología el 58,3% presentó pústulas subcórneas y en un porcentaje menor la formación de ampollas subepidérmicas (41,7%), mientras que el infiltrado inflamatorio de neutrófilos fue predominante en la totalidad de la muestra y un 25% presentó además la presencia de linfocitos. Con respecto a la IFD, clave para el diagnóstico, ninguno de los pacientes mostró depósitos granulares en la membrana basal, mientras que el 100% presentó cúmulos a nivel de las papilas dérmicas; datos similares a los encontrados por Fuertes29 y col, Alonso y col28 y Salmi y col30.
Los pacientes con DH presentaron una media de duración de la enfermedad de 6,6 meses (dato que no se corresponde a la totalidad de los pacientes, por pérdidas de registro), realizaron más tratamientos tópicos que sistémicos, aunque siempre asociados a una dieta estricta sin gluten, y presentaron en un 20% recaídas de la enfermedad. La duración de la enfermedad que determinaron Sobhan2 y col fue de 3,6 meses, con un porcentaje de recaídas del 18%. Fuertes29 y col no determinaron estos datos.
Pénfigo vulgar
Se trata de la tercera entidad en frecuencia de presentación de nuestra serie, con 9 casos (22,2%). En otras regiones en cambio, como demostraron los estudios iraníes,1,2, uno poblacional en Kuwait33 y otro en Grecia15 esta entidad representó un porcentaje superior al 80%; esto debido probablemente a las características étnicas de esas poblaciones. En el estudio prospectivo sobre incidencia de EAA alemán3 el 2% de los pacientes estudiados presentó diagnóstico de Pénfigo vulgar, mientras que el 14% de los pacientes del estudio retrospectivo realizado en el Reino Unido17 fue clasificado como esta entidad.
Es conocida la mayor frecuencia de presentación de esta entidad en judíos Ashkenaze, dato que no pudimos correlacionar en nuestro trabajo ya que, al ser retrospectivo, no contábamos con ese dato en la historia clínica.
La edad media de presentación fue de 49,2 años siendo, similar a la estimada por Rosti16 en su estudio en Rosario (50 años), Daneshpazhooh1 en Irán (43 años) y por Saha18 en el Reino Unido (49 años). El 77% de los casos perteneció al sexo femenino, aunque sin diferencias estadísticamente significativas, datos consistentes con la literatura.
Las manifestaciones clínicas predominantes en nuestro estudio fueron las erosiones en el 100% de los casos y solo 3 pacientes se presentaron ampollas fláccidas. La afectación mucosa se dio en el 54,54% de los pacientes, siendo en todos los casos la mucosa oral la afectada, hecho también descripto por Sobhan2 y col.
La histopatología y la IFD fueron similares en todos los pacientes: ampollas intraepidérmicas, sin un infiltrado inflamatorio predominante y depósitos interqueratinocitos de IgG.
Con respecto a las complicaciones durante el tratamiento, en este grupo de pacientes se presentaron infecciones, osteopenia y linfopenia (4 pacientes en total). Incluso tuvimos dos casos de muertes durante el tratamiento relacionadas con infecciones sistémicas y septicemia, lo que representó el 18% de los casos. Esto se condice con lo descripto en la bibliografía que estima un porcentaje variable de muertes en estos pacientes entre el 1 y 20%18,34.
Otras enfermedades ampollares autoinmunes
En nuestro trabajo se presentaron también otras enfermedades ampollares autoinmunes: Pénfigo foliáceo (1 caso), Dermatosis lineal por IgA (2 casos), Pénfigo por IgA (1 caso), Penfigoide de las mucosas (1 caso) y Lupus ampollar (1 caso), pero al ser tan escaso su porcentaje, no nos permitió el análisis estadístico de las mismas. Esto difiere con lo hallado en la literatura: Langan17 en su estudio multicéntrico poblacional en Alemania no halló casos de Pénfigo foliáceo, ni de Pénfigo por IgA, pero si describió 2 casos (que representaron el 5% en su estudio), de dermatosis lineal por IgA. En el Reino Unido17 no se encontraron casos con estos diagnósticos, mientras que en Irán1 se hallaron 0,2% de casos con Pénfigo por IgA, 4,4% de Pénfigo foliáceo y 10 casos (0,7%) de casos con Penfigoide de las mucosas.
A diferencia de otros estudios, en los que se describieron casos de Epidermólisis ampollar con una frecuencia de presentación dentro del grupo de las EAA del 0,5% para Irán1,2 y 2% para Kuwait33 y Alemania3, nosotros no tuvimos ningún caso que pudiéramos clasificar como tal.
CONCLUSIONES
Nuestros hallazgos permitieron definir que las EAA tienen una baja frecuencia de presentación (0,02%) siendo el PA la más frecuente, seguida por la DH y luego PV. Los hallazgos histopatológicos, de IFD y clínica se condicen con los hallados en la bibliografía, siendo los clásicos para cada enfermedad. Las comorbilidades más frecuentemente asociadas fueron la HTA, cardiopatía y demencia en el PA, enfermedad celíaca y autoinmunes (vitíligo, tiroiditis de Hashimoto) en la DH, mientras que para las otras EAA no se encontró asociación con comorbilidades. Los tratamientos realizados, a pesar de las diferencias entre las distintas EAA, fueron en su mayoría sistémicos con corticoides orales, asociados a azatioprina. No tuvimos pacientes con tratamiento biológico. Los tratamientos tópicos con corticoides representaron un alto porcentaje. El tiempo de duración de la enfermedad fue en promedio 13,04 meses 1mayor para los casos de Pénfigo vulgar. El control de la enfermedad se alcanzó en el 91% de los casos, mientras que el 78,75% de los pacientes logró la remisión completa. Si bien existieron recaídas en los pacientes estudiados (38,8%), nos fue imposible por el número de casos establecer una asociación entre estas y otros factores.
Si bien este trabajo representa un primer acercamiento para la caracterización epidemiológica de estos pacientes, se necesita de mayor profundización y estudios prospectivos para obtener información que permita un mejor manejo de estas patologías.

