











 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La IP, también conocida como síndrome de Bloch-Sulzberger, es una genodermatosis de herencia dominante ligada al cromosoma X, causada por una variante del gen IKBKG, que afecta principalmente a las mujeres y que suele ser fatal para los fetos masculinos.1,2Inicia, en la mayoría de los casos, en las dos primeras de semanas de vida.1Clínicamente, se caracteriza por lesiones cutáneas, localizadas clásicamente en extremidades, las cuales siguen las líneas de Blaschko y se desarrollan en cuatro fases clásicas: vesicular, verrugosa o hiperqueratósica, hiperpigmentada e hipopigmentada o atrófica.1El nombre de la patología deriva de los hallazgos histopatológicos típicos del tercer estadio, en el que se evidencia incontinencia de pigmento, depósito de melanina en la dermis papilar.1 Esta entidadse relaciona, además, con manifestaciones extracutáneas dentales, oftalmológicas y neurológicas,2entre otras, que suelen ser de aparición más tardía y de presentación variable.1Su pronóstico depende de la presencia y gravedad de estas, principalmente de las neurológicas, que incluyen convulsiones y retardo mental.3
El objetivo de esta publicación es comunicar un caso de incontinencia pigmenti, detallando el proceso diagnóstico, el seguimiento y la progresión de la dermatosis a través de los primeros tres estadios de la enfermedad-vesicular, verrugosa e hiperpigmentada-en el primer año de vida.
Caso clinico
La paciente es una niña de tres días de vida, con antecedente materno de aborto espontáneo de feto masculino. Fue evaluada en la sala de neonatología por el Servicio de Dermatología por presentar una dermatosis congénita localizada en tronco y miembros.
Al examen físico dermatológico presentaba lesiones lineales caracterizadas por múltiples pápulas eritematosas con erosión central secundaria al destechamiento de vesículas, y escasas vesículas y costras, sobre una base eritematosa. Estas se disponían siguiendo las líneas de Blaschko y se localizaban en miembros superiores e inferiores y tronco (Figuras 1 y 2). La paciente exhibía buen estado general y buena conducta alimentaria, y se encontraba afebril. No se objetivaron otros signos patológicos en el resto de la evaluación clínica.
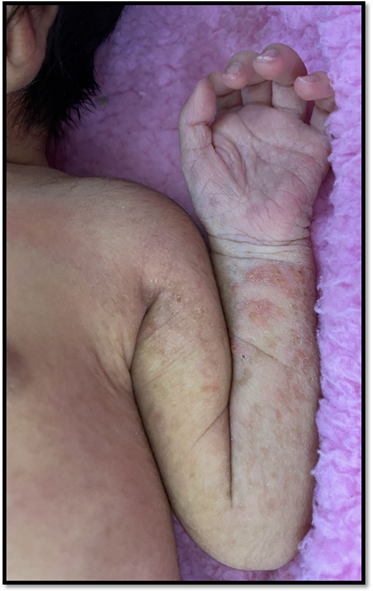
Figuras 1: Estadio I: lesiones lineales caracterizadas por múltiples pápulas eritematosas con erosión central secundaria al destechamiento de vesículas, y escasas vesículas y costras, que asientan sobre una base eritematosa, localizadas en miembros

Figuras 2: Estadio I: lesiones lineales caracterizadas por múltiples pápulas eritematosas con erosión central secundaria al destechamiento de vesículas, y escasas vesículas y costras, que asientan sobre una base eritematosa, localizadas en miembros
El diagnóstico presuntivo planteado fue incontinencia pigmenti, mientras que dentro de los diagnósticos diferenciales se consideraron herpes neonatal y mastocitosis congénita. Los estudios complementarios incluyeron laboratorio completo, toma del contenido de una vesícula para citodiagnóstico de Tzanck y una biopsia de piel para estudio histopatológico. Dentro de los signos de laboratorio considerados positivos se destaca la eosinofilia, mientras que en el citodiagnóstico sólo se observaron leucocitos polimorfonucleares.El estudio histopatológico informó epidermis con espongiosis eosinofílica y queratinocitos disqueratósicos, y un infiltrado inflamatorio dérmico predominantemente eosinofílico(Figuras 3 y Figuras 4).

Figuras 3: Estudio histopatológico: epidermis con espongiosis eosinofílica y queratinocitos disqueratósicos, y un infiltrado inflamatorio dérmico predominantemente eosinofílico (10x y 100x,hematoxilina y eosina).

Figuras 4: Estudio histopatológico: epidermis con espongiosis eosinofílica y queratinocitos disqueratósicos, y un infiltrado inflamatorio dérmico predominantemente eosinofílico (10x y 100x,hematoxilina y eosina).
De esta manera, con la clínica, el laboratorio y la histopatología, se arribó al diagnóstico de certeza de incontinencia pigmenti.
En consecuencia, se indicó tratamiento tópico con fomentos con agua de Alibour diluida y ácido fusídico al 2% en crema tres veces por día, con buena evolución de las lesiones.
Durante el seguimiento estricto de la paciente por nuestro Servicio se evidenció la evolución de la dermatosis. A los 17 días se observaron:I- en zona submentoniana, una lesión hiperpigmentada lineal pequeña con leve hiperqueratosis;II- en miembros, múltiples lesiones lineales caracterizadas por vesículas y costras melicéricas;III- en miembros inferiores, algunas lesiones hiperpigmentadas hiperqueratósicas(Figura 5),mientras que IV- en la vulva y en la raíz de miembros inferiores, se visualizaban eritema, múltiples vesículas y costras melicéricas(Figura6). Al mes siguiente, presentaba lesiones hiperpigmentadas lineales en miembros (Figura7). En miembros superiores y vulva persistían algunas pápulas eritematosas y vesículas (Figura8). En la última consulta del año, se evidenciaban solamente máculas hiperpigmentadas lineales que seguían las líneas de Blaschko localizadas en tronco y miembros (Figuras9 y 10). La paciente siempre demostró buen estado general y crecimiento normal.
En el transcurso de los controles se solicitó la evaluación por los servicios de neurología, oftalmología y cardiología, sin evidencia de signos patológicos. En la evaluación por odontología,en los iniciosde la dentición de la paciente se evidenció un diente cónico. Asimismo, la familia recibióconsejo genético sobre las implicancias de esta entidad, y en esta instancia se realizó un examen dermatológico a la madre, en quien no se constataron lesiones cutáneas sugestivas de la enfermedad.
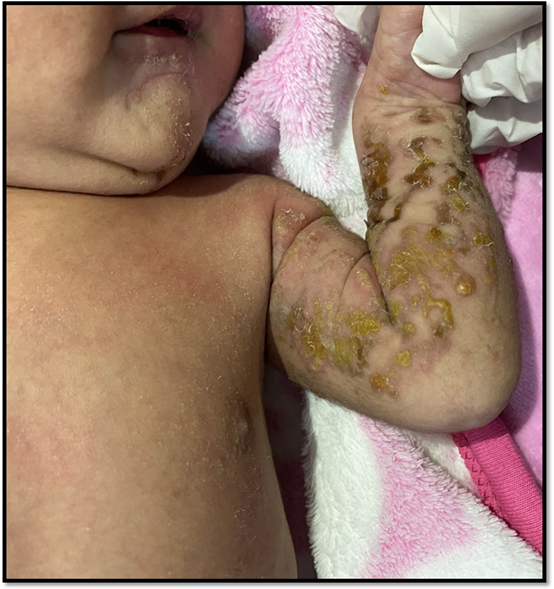
Figura 5: EstadioII: lesión hiperpigmentada lineal pequeña con leve hiperqueratosisen zona submentoniana, y múltiples lesiones lineales caracterizadas por vesículas y costras melicéricasen miembro superior izquierdo

Figura 6: EstadioII: eritema, múltiples vesículas y costras melicéricas en vulva y raíz de miembros inferiores


Figura 8: Estadio III: lesiones hiperpigmentadas lineales en miembros inferiores, y pápulas eritematosas y vesículas en vulva


Discusión
La incontinencia pigmenti es una genodermatosis infrecuente, sistémica, de herencia dominante ligada al cromosoma X, con penetrancia completa y expresividad variable.1,4Se define además como una displasia neuroectodérmica, ya que presenta malformaciones de diferentes tejidos y órganos derivados del ectodermo, que provocan alteraciones a nivel de la piel, los dientes, el sistema nervioso y los ojos, entre otras.1,4
Se origina por una variante genética con pérdida de función del IKBKG, previamente conocida como NEMO, ubicada en locus 28 del brazo largo del cromosoma X (Xq28),5 que codifica para la proteína IKK-gamma.6 Ésta modula la actividad del factor de transcripción nuclear kappa B (NF-B) que interviene en la regulación del sistema inmunológico, particularmente en los mecanismos de apoptosis celular inducida por el factor de necrosis tumoral alfa (TNF-), con lo que provoca una alteración en el desarrollo ectodérmico.6,7
La incidencia es de aproximadamente 0,7 casos cada 100.000 nacidos vivos,8 y su prevalencia no se conoce con precisión,9 dado que es una patología infradiagnosticada.3,10Entre el 65 a 75% se deben a mutaciones esporádicas o “de novo”.1 Predomina en las mujeres con una relación 35:1,11 ya que en fetos masculinos es usualmente letal intrauterino. Sin embargo, existen casos de hombres afectados debido a la presencia de anomalías cromosómicas (síndrome de Klinefelter, 47XXY), mosaicismos somáticos o mutaciones hipomorfas.10,11Las mujeres sobreviven debido al fenómeno de lionización, que es la inactivación selectiva de uno de los cromosomas X-un mosaicismo funcional-durante la embriogénesis.1,5
Clínicamente, se caracteriza por lesiones cutáneas que siguen las líneas de Blaschko y que se desarrollan en cuatro estadios con una secuencia cronológica característica: vesicular o inflamatoria, verrugosa o hiperqueratósica, hiperpigmentada e hipopigmentada o atrófica.1,3Cabe destacar que estas etapas pueden estar superpuestas; alguna, incluso, puede estar ausente, ademásde observarse diferencias tanto en su inicio como en su duración.1,3
El primer estadio, el vesicular, que se presenta en el 90-95% de los casos, se caracteriza por el desarrollo de pápulas, vesículas o pústulas sobre una base eritematosa, distribuidas principalmente en extremidades, pero también en tronco, cabeza y cuello.12 Están presentes desde el nacimiento o aparecen durante las primeras dos semanas de vida,e involucionanluego de cuatro a seis meses.4
La segunda etapa, la verrugosa, presente en el 70-80% de los pacientes, se caracteriza por pápulas y placas verrugosas o hiperqueratósicas lineales que se desarrollan en extremidades y tronco. Se presentan desde las dos a seis semanas de vida, se resuelvena los seis meses aproximadamente.4,12
El tercer estadio, el hiperpigmentado, el más frecuente (98%), muestra máculas hiperpigmentadas grisáceas o amarronadas, lineales o circulares, que comprometen tronco, extremidades y pliegues. Estas lesiones parecen no tener relación con las anteriores, pueden aparecer al mes de vida y generalmente remiten durante la adolescencia.4,12
En el último estadio, la hipopigmentada, la más inconstante (30-75%), se visualizan lesiones maculares hipopigmentadas y atróficas con ausencia de folículos, localizadas en extremidades superiores e inferiores. Usualmente se desarrollan durante la pubertad, y son permanentes.4,12
Se han descripto reactivaciones de las lesiones cutáneas, principalmente lasdel estadiovesicular, luego de la exposición a ciertos factores desencadenantes, como infecciones, fiebre, vacunas y tratamientos conláser.13,14
A pesar de que existe una importante heterogeneidad en la clínica de la incontinencia pigmenti, las lesiones cutáneas son el primer signo clínico en prácticamente todos los casos.5
Además, pueden verse afectados los anexos cutáneos: el pelo en un 28 a 38% de los casos y las uñas en un 17 a 34%.5Los signos incluyen alopecia difusa o de tipo cicatricial que afecta principalmente la zona del vértex, pero que también puede comprometer cejas, pestañas y vello corporal. En cuanto a las alteraciones ungueales, se describen desde estrías longitudinales y hoyuelos hasta distrofia ungueal y tumores ungueales, en la mayoría o todas las uñas de las manos, más frecuentemente, yen lasde los pies. Estas, generalmente, se desarrollan más tardíamente.15,16
Los signos dermatológicos se relacionan con diferentes manifestaciones extracutáneas. Las más frecuentes son las odontológicas (80%),3 neurológicas (30%)17 y las oculares (20-37%),17que suelen ser de aparición más tardía, yque demuestranuna heterogeneidad importante en los diferentes pacientes.16
Las anomalías dentales (anodoncia, dentición retrasada, dientes cónicos y microdoncia) son importantes para arribar al diagnóstico, ya que, a diferencia de las manifestaciones cutáneas, persisten toda la vida, perose manifiestan recién al año con la dentición.5,10
Se destaca la importancia de una precoz derivación a oftalmología a fin de comprobar la existencia, o no, de alteraciones oftalmológicas, especialmente las retinianas, que son las más frecuentes, debido a que puede generar una morbilidad significativa, que puede llevar a la ceguera.16Estas incluyen telangiectasias, ectasias, hemorragias, anastomosis arteriovenosas, neovascularización y avascularización, que pueden derivar en desprendimiento de la retina.16Además, se describen anomalías no-retinianas, como cataratas, microftalmia,estrabismo, hipermetropía y miopía, entre otras.16
Las anomalías del sistema nervioso central, aunque menos frecuentes, son las más graves y se relacionan en muchos casos con las oftalmológicas.2 Comprenden crisis convulsivas, disfunción motora, retardo mental y microcefalia.7 Algunas de estas pueden correlacionarse con signos-atrofia cerebral y cerebelosa-en los estudios de imágenes, particularmente la resonancia magnética nuclear con técnica de difusión.18
Asimismo, en estos pacientes se han reportado una mayor cantidad de anomalías mamarias y de los pezones, comopolitelia, hipoplasia del pezón y aplasia e hipoplasia mamaria, y alteraciones palatinas.5,6En raras ocasiones, se han reportado anomalías cardiovasculares e hipertensión pulmonar en niños con esta enfermedad.19
Aunque el diagnóstico es clínico, se puede complementar con algunos estudios complementarios.15 La realización de una biopsia cutánea es de gran utilidad. Cada estadioclínica tiene signos histopatológicos particulares.2,3En el primer estadio, el vesicular, se describen espongiosis eosinofílica, vesículas intraepidérmicas que contienen eosinófilos y apoptosis de queratinocitos en la epidermis.2,3El siguiente estadio, el hiperqueratósico, presenta papilomatosis e hiperqueratosis, acantosis y disqueratosis en la epidermis.2,3El tercer estadio, el hiperpigmentado, se caracteriza por una importante incontinencia de pigmento melánico y numerosos melanófagos en la dermis,2,3 lo que le confiere el nombre a esta genodermatosis. En el último estadio, se observan atrofia epidérmica, disminución importante de melanina en la capa basal, cuerpos apoptóticos en la epidermis y la dermis papilar, y ausencia de anexos cutáneos.2,3 Además, el hallazgo de eosinofilia en el laboratorio es característico.20 Por último, se puede realizar el estudio genético para pesquisar la variante genética tomando una muestra de sangre periférica del paciente.16
Con el propósito de facilitar el diagnóstico de esta entidad, se han propuesto una serie de criterios (Tabla 1)21 descriptos en 1993, revisados en el 2014(Tabla 2)16y actualizados en el 2020 (Tabla 3),22 que incluyen los signos clínicos, los estudios complementarios y el estudio genético. Según la última actualización, se consideran como criterios principales las lesiones cutáneas típicas de cada estadio siguiendo las líneas de Blaschko, las anomalías dentalesy el estudio genético con la identificación del reordenamientodel gen IKBKG. Los secundarios incluyen los diferentes signos del pelo, de las uñas, de las glándulas mamarias y de la retina característicos de este trastorno, la eosinofiliaen el estadio 1 y la histología cutánea característica.Se arriba al diagnóstico de incontinencia pigmenticon la presencia de1 criterio secundario si existe un familiar de primer grado de sexo femenino afectado y con 1 criterio principal en el caso de ausencia de antecedentes familiares. Se debe dudar del diagnóstico si no se observa ningún criterio secundario.22
Los diagnósticos diferenciales comprenden cualquier afección con lesiones que siguen líneas de Blaschko, y dependen del estadio de incontinencia pigmenti (Tabla 4).1,12Se debe considerar siempre la edad del paciente y el momento del comienzo de la dermatosis.16
La asistencia de esta entidad es multidisciplinaria.15 El paciente debe tener una evaluación inicial temprana y un seguimiento anual con los servicios de dermatología, oftalmología, neurología, odontología y genética.1 Las lesiones cutáneas generalmente no requieren tratamiento.3 No obstante, se ha reportado el uso de corticoides de alta potencia (diflucortolona)23 e inhibidores de la calcineurinatópicos (tacrolimus 0,1%)24que evitan la progresión de las manifestaciones en piel. En cuanto a los pacientes con síntomas neurológicos y oftalmológicos, se recomienda la realización de neuroimágenes.17 Debido a que por el momento no existe un tratamiento específico que cambie el curso natural de la enfermedad, se tratan los síntomas, las complicaciones y las comorbilidades.3 La morbilidad y el pronóstico dependen de la presencia y gravedad de estas, principalmente de las oftalmológicas y neurológicas,8 que en muchos casos determinan la calidad de vida del paciente y que en ocasiones son causa de muerte.
Se debe, además, evaluar a los familiares de primer grado de género femenino para detectar la enfermedad. El asesoramiento genético es fundamental, y se debe informar a los pacientes de la alta posibilidad de abortos, particularmente de embriones masculinos.5,15

Tabla 1: Criterios diagnósticos de incontinencia pigmentiadaptado de S J Landy y D Donnai (1993)21

Tabla 2: Criterios diagnósticos principales y secundarios de la incontinencia pigmentiadaptado de Minić S, Trpinac D, Obradović M (2014)16

Tabla 3: Criterios diagnósticos actualizados para incontinencia pigmenti a partir de los criterios de Landy y Donnai. C. Bodemer y col. (2020)22

Tabla 4: Diagnósticos diferencialessegún cada estadio12
Conclusión
Pese a su infrecuencia, se remarca la importancia del conocimiento de la incontinencia pigmenti, particularmente por dermatólogos y pediatras, ya que los hallazgos dermatológicos suelen ser los primeros en manifestarse y a edades muy tempranas, con lo que se constituyen en el principal criterio diagnóstico. Los estudios complementarios, particularmente la biopsia de piel, son herramientas importantes que se deben implementar para apoyar el diagnóstico clínico, y siempre que se encuentre disponible, se recomienda la realización del estudio genético para certificar la sospecha clínica. La paciente del caso descripto presentaba los cuatro criterios mayores, dos criterios menores y eosinofilia. El diagnóstico temprano y el seguimiento multidisciplinario para advertir precozmente las posibles complicaciones extracutáneas permiten una mejor calidad de vida en estos pacientes. El tratamiento de las complicaciones oftalmológicas puede prevenir el desarrollo de ceguera y el de las complicaciones neurológicas, secuelas importantes e incluso la muerte.
Recibido: 10/02/2023
Recibido 1°Corrector: 06/05/2023
Recibido 2° corrector: 18/11/2023
Aceptado para su Publicación: 26/11/2023

