










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de endocrinología y metabolismo
versión On-line ISSN 1851-3034
Rev. argent. endocrinol. metab. v.45 n.2 Ciudad Autónoma de Buenos Aires abr./jun. 2008
Alteraciones neuroendocrinas del síndrome de poliquistosis ovárica en la adolescencia
Neuroendrocrine anormalities in adolescents with polycystiz ovary sindrome
Ropelato M. G.*
* MG Ropelato es miembro de la Carrera de Investigador del Consejo de Investigación en Salud del Gobierno de la Ciudad Autónoma de Buenos Aires.
División de Endocrinología, Centro de Investigaciones Endocrinológicas (CEDIE), Hospital de Niños Dr. Ricardo Gutiérrez. Buenos Aires, Argentina.
Dirección Postal: Gallo 1360. (1425) Ciudad Autónoma de Buenos Aires.
E-mail: gropelato@cedie.org.ar
Correspondencia a telefax: (5411) 4963-5931 ext 131 / (5411) 4963-5930
Resumen
El síndrome de poliquistosis ovárica (SPCO) es una de las endocrinopatías más comunes que afecta a las mujeres en edad reproductiva, su expresión clínica comienza en edad perimenárquica y si bien fue descripto hace más de 70 años, hasta el presente, el(los) mecanismo(s) fisiopatológico(s) que lo origina(n) no se conoce(n) con certeza.
Debido a la gran heterogeneidad en la expresión clínica y bioquímica que caracteriza al SPCO es probable que existan subgrupos de pacientes en las que sea posible identificar alguno de los mecanismos implicados en la patogenia como el responsable de los principales signos y síntomas observados. La presente revisión propone conocer en profundidad las anormalidades neuroendocrinas como uno de los principales componentes del síndrome.
En nuestra experiencia, las adolescentes con SPCO presentan hipersecreción de LH (aumento de la masa de LH secretada por pulso, de la frecuencia de pulsos y de la tasa de producción), y un patrón desordenado de secreción de LH (mayores valores de ApEn) en relación a adolescentes eumenorreicas. Varias líneas de evidencia sugieren que uno de los mecanismos responsables de estos defectos es el aumento de frecuencia de secreción del GnRH. Las adolescentes con SPCO secretan moléculas de LH con mayor actividad biológica y mayor proporción de isoformas con punto isoeléctrico más alcalino que las adolescentes eumenorreicas. La preponderancia de isoformas más básicas y más bioactivas en estas pacientes se relaciona con elevados niveles séricos de 17-hidroxiprogesterona, androstenodiona (A) y testosterona (T). El aumento de la frecuencia de pulsos de GnRH y un microambiente hormonal caracterizado por exceso de andrógenos podrían conjuntamente promover la predominante secreción de este tipo de isoformas de LH.
En ausencia de obesidad, las pacientes con SPCO presentan un incremento de la tasa de producción de GH y un patrón de secreción más ordenado (menores valores de ApEn, similar al patrón de secreción de GH observado en el varón adulto). La mayor secreción de GH podría potenciar la acción gonadotrófica sobre la esteroideogénesis ovárica.
Analizando la sincronía entre pares de hormonas relacionadas mediante dos técnicas complementarias (cross ApEn y cross correlación) se demuestra que las adolescentes con SPCO presentan un deterioro en las asociaciones entre LH-andrógenos comparadas con las adolescentes eumenorreicas.
El desacople de la secreción bihormonal (LH-A y LH-T) en adolescentes con SPCO es consistente con defectos en el control de la secreción ovárica de andrógenos dependiente de LH y con una alteración en el control negativo que ejercen los andrógenos sobre la secreción GnRH/LH. Estas alteraciones neuroendocrinas en la unidad GnRH/LH y andrógenos ováricos podrían promover el hiperandrogenismo y alterar la maduración folicular.
Palabras clave: Síndrome de poliquistosis ovárica; Adolescencia; LH; FSH; Andrógenos
Abstract
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder among women in reproductive age, frequently begins during adolescence causing menstrual irregularity and hirsutism. Although described up more than seventy years ago, the primary pathophysiologic mechanisms underlying this disorder remain unknown.There is not a single etiologic factor that fully accounts for the spectrum of abnormalities in the PCOS. This review addresses current knowledge about the neuroendocrine abnormalities as a major component of the syndrome.
From this perspective, adolescents with PCOS exhibit an accelerated frequency and/or higher amplitude of LH pulses, augmentation of secretory burst mass, and a more disorderly LH release (higher ApEn) than eumenorrheic adolescents. Several lines of evidence suggest that the mechanisms underlying the defects in LH secretion in PCOS include an increased frequency of GnRH secretion. These patients also show elevated in vitro LH bioactivity and a preponderance of basic LH isoforms, which correlate positively with elevated serum of 17-hydroxyprogesterone, androstenedione (A), and testosterone (T) concentrations. Heightened GnRH drive of gonadotropin secretion and steroid-permissive milieu appear to jointly promote elevated secretion of basic LH isoforms.
Non obese adolescents with PCOS secrete GH at a higher rate and with more orderly patterns (resembling a male profile) than controls. Indeed, GH appears to act as a co-gonadotropin.
When synchronicity of paired hormone profiles was appraised by two independent, but complementary, statistical tools (cross-entropy and cross correlation), concomitant uncoupling of the pairwise synchrony of LH - androgens was demonstrated in girls with PCOS. Asynchrony of LH-A and LH-T pairs further localizes a pathway defect to LH-dependent feedforward control of ovarian androgen secretion. These abnormalities are also consistent with altered androgen negative feed-back regulation of GnRH/LH output.
These data suggest that in PCOS there are anomalies of signaling between GnRH/LH and ovarian androgens that promote hiperandrogenism and impaired follicle maturation.
Key Words: Polycystic ovary syndrome; LH; FSH; Androgen
Introdución
El síndrome de poliquistosis ovárica (SPCO) afecta aproximadamente a un 5 % de las mujeres en edad reproductiva, presenta un comienzo perimenárquico y causa alteraciones en el eje reproductivo que incluyen oligo- o anovulación junto con evidencias clínicas y/o bioquímicas de hiperandrogenismo(1-3). Se acompaña de trastornos de la fertilidad, aumento de abortos espontáneos, mayor riesgo de cáncer de endometrio, alteraciones del metabolismo (resistencia a la insulina y dislipidemia) y riesgo de desarrollar Diabetes Mellitus Tipo II en la vida adulta(4,5).
La primera descripción del síndrome fue realizada en 1935 por Stein y Leventhal(6), quienes asociaron un cuadro clínico caracterizado por amenorrea u oligomenorrea, hirsutismo y obesidad a los ovarios poliquísticos. Desde ese momento, los estudios morfológicos e histológicos de los ovarios de estas pacientes demostraron engrosamiento de la albugínea, hiperplasia de la teca interna y del estroma cortical y presencia de múltiples folículos subcapsulares en diversos estadios de atresia.
En 1964, Stein demostró que la resección en cuña bilateral de los ovarios de mujeres adultas con la enfermedad provocaba la reanudación de ciclos menstruales y en algunos casos la concepción(7). Sobre la base de estos resultados se pensó en un defecto primario del ovario y se le asignó el nombre de enfermedad del ovario poliquístico. El reconocimiento de la heterogeneidad tanto de la histología ovárica como de los hallazgos clínicos y bioquímicos en las mujeres afectadas promovió el uso del término de síndrome de poliquistosis ovárica (SPCO).
Si bien este síndrome fue descripto hace más de 70 años, hasta el presente no se conoce su etiología.
Los mecanismos involucrados en la patogénesis del SPCO aún no se han demostrado con certeza, sin embargo existen evidencias que indican alteraciones en el control endocrino, paracrino y autocrino del proceso de foliculogénesis. El conocimiento de algunas de estas alteraciones ha llevado a postular varias hipótesis tendientes a explicar el origen del síndrome y que no son excluyentes(8):
• Anormalidades neuroendocrinas en la secreción de gonadotrofinas: Un defecto neuroendocrino primario en el generador de pulsos de GnRH, lleva a la secreción exagerada de LH, aumento en la producción de andrógenos por el ovario y anovulación.
• Anormalidades en la síntesis de esteroides en el ovario: Un defecto primario en la síntesis de andrógenos por el ovario resulta en hiperandrogenismo y anovulación.
• Anormalidades metabólicas: Defectos en la acción de la insulina (resistencia a la insulina) se acompañan de hiperinsulinismo y esto induce un exceso en la secreción de andrógenos y anovulación.
• Alteraciones en los mecanismos paracrinos y autocrinos de la foliculogénesis: La detención del desarrollo folicular que provoca la anovulación se debería a anormalidades en los sistemas intraováricos que coregulan el desarrollo y la maduración folicular.
• Causas hereditarias: El síndrome de poliquistosis ovárica es un desorden multigénico complejo y los genes candidatos son los que intervienen en la regulación del eje hipotalamo- hipofiso -ovárico.
En esta revisión se explora la hipótesis de que anormalidades neuroendocrinas puedan ser la base fisiopatológica del síndrome produciendo hipersecreción de LH, aumento de la producción de andrógenos por el ovario y anovulación.
El estudio de las alteraciones neuroendocrinas en adolescentes con SPCO podría revelar los mecanismos fisiopatológicos en las etapas iniciales del síndrome sin que estos resultados sean enmascarados por las consecuencias del mismo.
Definición del síndrome de poliquistosis ovárica
Hasta el momento han existido dos definiciones del SPCO. La primera proviene de la publicación de una conferencia de expertos del Instituto Nacional de la Salud de los EE.UU. sobre SPCO en abril de 1990 (Criterios del NIH, 1990), y propone tres criterios mayores (en orden de importancia)(9): 1) hiperandrogenismo y/o hiperandrogenemia; 2) oligo- o anovulación y 3) exclusión de otras enfermedades que cursan con exceso de andrógenos: hiperplasia suprarrenal congénita, hiperprolactinemia, síndrome de Cushing, disfunción tiroidea (Ver tabla 1). La presencia de ovarios poliquísticos por ecografía se consideró un criterio de gran controversia. En esencia, los resultados de esta conferencia de expertos identificaron al SPCO como un desorden de exceso de andrógenos.
Tabla 1: Entidades clínicas que deben ser excluidas en el diagnóstico diferencial del síndrome de poliquistosis ovárica (Modificado de Ref. 8).
La segunda definición proviene de otra conferencia de expertos organizada por ESHRE/ASRM en Rotterdam en mayo de 2003 (Criterios de Rotterdam, 2003)(10,11). Este consenso determinó que el síndrome debe ser diagnosticado, después de la exclusión de otras enfermedades relacionadas (ver Tabla 1), por dos de los siguientes tres criterios: 1) oligo o anovulación, 2) signos clínicos y/o bioquímicos de hiperandrogenismo y 3) ovarios poliquísticos por ecografía.
La definición de SPCO de Rotterdam 2003, expande la definición del NIH 1990 a dos nuevos fenotipos de SPCO incluyendo a mujeres con: 1) ovarios poliquísticos y signos clínicos y/o bioquímicos de exceso de andrógenos pero sin anovulación y 2) ovarios poliquísticos y anovulación pero sin hiperandrogenemia y/o hirsutismo (es decir sin signos de exceso de andrógenos). La inclusión de los ovarios poliquísticos como criterio ha originado un importante debate y recientemente se han publicado posiciones en contra(12) y a favor(13) de la definición de Rotterdam. Para conciliar estas posiciones y determinar los criterios que definen al síndrome, la Sociedad de Exceso de Andrógenos (AES) encomendó a un grupo internacional de expertos la revisión sistemática de trabajos de investigación sobre los aspectos epidemiológicos y fenotípicos del síndrome con el fin de guiar el diagnóstico clínico y las futuras investigaciones sobre SPCO. Así, se publicó la última definición de SPCO(14) que determina que los criterios son los siguientes: 1) hiperandrogenismo: hirsutismo y/o hiperandrogenemia, 2) disfunción ovárica: oligo- anovulación y/o ovarios poliquísticos por ecografía y 3) exclusión de otros desórdenes de andrógenos o enfermedades relacionadas. La aplicación de esta última definición identifica 9 posibles fenotipos siendo el exceso de andrógenos una característica principal y excluye el fenotipo de ovarios poliquísticos y anovulación sin hiperandrogenemia y/o hirsutismo.
Características clínicas, bioquímicas y ecográficas del síndrome
Las principales alteraciones que se presentan en mujeres que sufren este síndrome son anovulación, hiperandrogenismo y obesidad.
El 70 % de las adolescentes con SPCO presenta irregularidades menstruales como manifestación de la anovulación u oligoovulación crónica. Luego de al menos dos años posmenarca, la insuficiencia ovulatoria se expresa típicamente como oligomenorrea (menos de 9 ciclos menstruales por año) o amenorrea (Tabla 2). Algunas adolescentes con SPCO pueden presentar amenorrea primaria (ausencia de menarca a los 16 años de edad) o amenorrea secundaria (ausencia de menstruaciones por al menos tres meses). Los ciclos anovulatorios pueden, también, producir hemorragia uterina disfuncional, y por último, en algunos casos, las pacientes pueden presentar ciclos menstruales de características normales.
Tabla 2: Signos y síntomas clínicos, alteraciones hormonales y criterios ecográficos para el diagnóstico del SPCO.
Las manifestaciones cutáneas del hiperandrogenismo incluyen hirsutismo, acné, y alopecía androgénica pero también existen formas crípticas en las que no se observan estas manifestaciones a pesar del aumento en el nivel de andrógenos circulantes.
Es ampliamente reconocido que el SPCO es en esencia un síndrome de exceso de andrógenos en la mujer. Teóricamente, la determinación de testosterona (T) total y T libre en suero en fase folicular temprana serían de ayuda en la confirmación del diagnóstico del SPCO, especialmente en ausencia de signos clínicos del exceso de andrógenos. Sin embargo, la determinación de la concentración sérica de testosterona total presenta serias dificultades:
• La T (al igual que otros andrógenos) presenta cambios pulsátiles, diurnos y cíclicos(15)
• La mayoría de los laboratorios proveen intervalos de referencia excesivamente amplios, dado que la población general incluye mujeres con exceso de andrógenos moderado y asintomático(16)
• La mayoría de los ensayos comerciales son directos (sin extracción previa del esteroide) e inexactos(17)
En general, un nivel de T de 0.90 ng/ml o mayor (confirmado), resulta buena evidencia del exceso de andrógenos, si es superior a los 2 ng/ml el diagnóstico de neoplasia productora de andrógenos es altamente probable (Tabla 1).
La concentración de T libre en fase folicular temprana (FFT) del ciclo menstrual puede ser elevada, aún cuando la T total sea normal, y es 50 % más sensible que la T total en el diagnóstico del exceso de andrógenos. Sin embargo, el método de referencia para medir T libre no está disponible en los laboratorios de rutina y los ensayos comerciales no son exactos. Además, los valores de referencia no se encuentran bien establecidos, especialmente para adolescentes.
La obesidad es común en pacientes con SPCO, sin embargo no puede considerarse como un prerrequisito para el diagnóstico, ya que el 50 % de las mujeres con síndrome no son obesas(1).
Las pacientes con SPCO presentan obesidad de tipo androide (adiposidad central con circunferencia de cintura > a 88 cm una vez que se completó el desarrollo puberal). La obesidad y presencia de acantosis nigricans (un signo de insulinorresistencia) pueden asociarse en estas pacientes.
Si bien la obesidad no puede considerarse un evento iniciador en el desarrollo del síndrome, su presencia puede modificar el patrón hormonal y producir disturbios metabólicos.
Las alteraciones ecográficas del ovario asociadas al síndrome no son patognomónicas dado que pueden observarse en otras condiciones patológicas como hiperprolactinemia, disfunción tiroidea, síndrome de Cushing, e hiperplasia suprarrenal congénita(2). Más aún, el 20 % de la población femenina con ciclos menstruales ovulatorios presenta un fenotipo de ovarios multiquísticos en estudios ecográficos(18). Los signos ecográficos de ovarios poliquísticos son menos demostrables en adolescentes respecto de adultas con el síndrome probablemente porque los estudios se realizan por vía abdominal en lugar de la vía transvaginal(19).
El ovario poliquístico se define por un volumen ovárico > 10 cm3 o aquel que contiene 10 o más folículos de 2 a 9 mm de diámetro (microquistes,(20) (Tabla 2). Este criterio asume la ausencia de un folículo dominante (> a 10 mm de diámetro) o de cuerpo lúteo. El aumento del volumen ovárico es un indicador objetivo del incremento del estroma ovárico. En las adolescentes, el aumento del volumen ovárico por encima de 10 cm3 define la presencia de ovarios poliquísticos(21) (Tabla 2). Otro criterio propone sumar a los signos ecográficos anteriores un incremento del estroma superior al 25 % del volumen ovárico(22). Un trabajo más reciente define al ovario poliquístico cuando la relación área del estroma/área total del ovario es superior a 0.34(23).
Los síntomas del síndrome de ovario poliquístico generalmente comienzan a edad perimenárquica(24). Sin embargo, el síndrome también puede presentarse después de la pubertad como resultado de modificadores externos (por ejemplo la obesidad).
La pubarca precoz, que es el resultado de un aumento de secreción de andrógenos adrenales antes de la edad en la que se produce normalmente la adrenarca, es un signo que se asocia frecuentemente con aparición de SPCO luego de la menarca(25). Recientemente, se ha postulado que el retraso de crecimiento intrauterino podría ser un factor etiopatogénico del síndrome de poliquistosis ovárica, particularmente en sus componentes metabólicos(26).
La pubertad precoz central idiopática (PPCI) se ha considerado una condición benigna, sin consecuencias serias en la vida adulta. Sin embargo, algunas niñas desarrollan un cuadro de hiperandrogenismo similar al observado en adolescentes con SPCO. Recientemente, hemos demostrado hipersecreción de LH en adolescentes hiperandrogénicas con historia de PPCI(27). Estos hallazgos permitirían hipotetizar la existencia de un mecanismo común (alteración de la actividad del generador de pulsos de GnRH) subyacente causante de los cuadros de PPC y SPCO en estas jóvenes.
Se ha demostrado que el perfil secretorio de gonadotrofinas, caracterizado por secreción predominante de LH, que se observa en mujeres adultas con el síndrome de poliquistosis ovárica, semeja las características fisiológicas de adolescentes perimenárquicas. La anovulación así como el aumento moderado de los niveles plasmáticos de LH y andrógenos son una característica de los primeros ciclos posteriores a la menarca(28).
La instauración del mecanismo de “feedback” positivo, el último evento que refleja una funcionalidad completa del eje gonadal y que asegura la ovulación, se alcanza luego que el patrón de secreción de gonadotrofinas pierde el ritmo circadiano(29). Este mecanismo se desarrolla varios años después de la menarca(30). Se ha sugerido que las irregularidades menstruales que presentan las adolescentes en los primeros años posteriores a la menarca podrían ser el resultado de una inmadurez en el mecanismo de “feedback” positivo. Varios estudios utilizando RIA han demostrado que la ritmicidad de la secreción de gonadotrofinas está comprometida en las adolescentes que persisten con ciclos anovulatorios después de 2 años posteriores a la menarca(28,30). En niñas hiperandrogénicas en diversos estadios del desarrollo puberal, se demostró la existencia de un ritmo circadiano invertido en la secreción de LH con incremento de los pulsos de LH durante el día y se postuló que la persistencia de estas alteraciones neuroendocrinas marcaban la evolución a un SPCO en la vida adulta(31,32).
Las adolescentes que persisten con ciclos anovulatorios presentan muy altos niveles de andrógenos en asociación con elevados niveles de LH que provocarían una inadecuada progresión y maduración folicular, desarrollándose folículos que van a la atresia y que son incapaces de generar el ritmo cíclico de secreción de esteroides que caracteriza a la ovulación(30).
Evidencias de anormalidades neuroendocrinas en adolescentes con SPCO
La patogénesis del síndrome de poliquistosis ovárica es objeto de gran controversia y debate. La heterogeneidad clínica y bioquímica de este desorden sumado a la falta de un modelo animal adecuado para el estudio del mismo hacen muy difícil establecer su etiopatogenia.
A continuación se resumen y discuten las principales anormalidades neuroendocrinas en adolescentes con el síndrome de poliquistosis ovárica: las características del patrón de secreción de gonadotrofinas y su interrelación con el sistema de la hormona de crecimiento (GH) y con factores reguladores intraováricos (inhibinas, IGFs, IGFsBPs).
1. Hipersecreción de LH (inmuno- y bioactiva) y de isoformas más alcalinas de LH
Los primeros estudios de pulsatilidad de gonadotrofinas en mujeres adultas con SPCO utilizando RIA y métodos discretos para el análisis de pulsos demostraron que los altos niveles circulantes de LH se mantienen por una exagerada descarga pulsátil de LH, especialmente a través del aumento de
la amplitud y de la frecuencia de pulsos(33-35).
Recientemente, hemos estudiado las características de secreción de LH y de FSH de 11 adolescentes con SPCO y 10 adolescentes eumenorreicas con similar edad cronológica, ginecológica e índice de masa corporal(36). Se determinó el patrón de secreción de gonadotrofinas combinando inmunoensayos fluorométricos (IFMAs) altamente sensibles y específicos y análisis de deconvolución. Las principales alteraciones en la secreción de LH se resumen en la Figura 1. El aumento de la secreción de LH ocurrió a expensas de incremento de la tasa de secreción pulsátil de LH (aumento de la masa secretada por pulso y de la frecuencia de eventos secretorios) y de la tasa de secreción basal de LH. Para cuantificar la regularidad del perfil secretorio de LH se calculó la entropía aproximada (ApEn). Como muestra la Figura 1 las pacientes SPCO presentaron un patrón de secreción de LH marcadamente desordenado (mayores valores de ApEn).
Figura 1: Parámetros de la secreción de LH (IFMA) obtenidos por deconvolución en adolescentes con SPCO y controles eumenorreicas. La línea horizontal representa la media para cada grupo.
El aumento de LH (masa de LH liberada por pulso y tasa de producción) se relacionó en forma positiva y significativa con los elevados niveles de 17-OH progesterona y androstenodiona. Otros autores(37,38) han demostrado asociación positiva entre la media de la secreción de LH y 17-OH progesterona y androstenodiona en niñas hiperandrogénicas y adultas con SPCO. La elevación conjunta de LH y andrógenos ováricos sugiere un defecto en el mecanismo de feed-back negativo.
Se ha investigado la relación entre la obesidad y la inapropiada secreción de gonadotrofinas en SPCO(37-40). En concordancia con otros estudios en adultas(38,39), observamos en adolescentes con SPCO, una influencia negativa del IMC sobre la media de secreción de LH, disminuyendo específicamente la masa de LH liberada por pulso y por lo tanto de la tasa de producción pulsátil de LH. Se ha propuesto que algún factor asociado con el aumento de la masa grasa podría suprimir la amplitud de pulsos de LH inducida por GnRH o la capacidad de respuesta del gonadotropo al GnRH pero no la frecuencia de secreción de LH en este síndrome(38,39). Arroyo y col.(39) demostraron en mujeres adultas con SPCO que las concentraciones de LH y la amplitud de los pulsos se relacionaron inversamente con la concentración integrada de insulina en 24 hs. y el grado de resistencia a la insulina. Otro posible candidato es la leptina, sin embargo, la mayoría de los estudios publicados sobre este péptido y SPCO no encontraron relación entre los niveles basales de leptina y LH(40-44). Dado que normalmente la secreción de leptina se acopla minuto a minuto con la secreción de LH(45) es posible que ocurran alteraciones en este acoplamiento en el síndrome de poliquistosis ovárica que no se detecten con muestras basales.
Los datos presentados hasta aquí aportan evidencias sobre las alteraciones en la secreción de LH inmunoactiva en las etapas iniciales del síndrome de poliquistosis ovárica. Es escasa la información en la literatura sobre la posible relación entre las características de la secreción de LH y su actividad biológica, así como el tipo de isoformas de LH que acompañan a estas anormalidades secretorias.
Basados en un muestreo nocturno espontáneo demostramos que las adolescentes con SPCO presentan niveles de LH biológicamente activa significativamente mayores a las del grupo control (Figura 2). La separación por cromatoenfoque de las isoformas de LH presentes en las muestras de suero de las pacientes con SPCO demostró la existencia de mayor cantidad de LH en los intervalos de pH más básicos (PH > 8 y pH: 7.99 - 7.0) comparadas con las adolescentes eumenorreicas (p=0.031, Figura 3)(46). Estos hallazgos son coincidentes con lo observado por estudios previos en adultas con el síndrome(47,48). Sin embargo, nuestros datos aportaron evidencias por primera vez sobre la asociación entre la hipersecreción pulsátil de LH inmunoactiva y la secreción de isoformas más bioactivas in vitro y con punto isoeléctrico más alcalino.
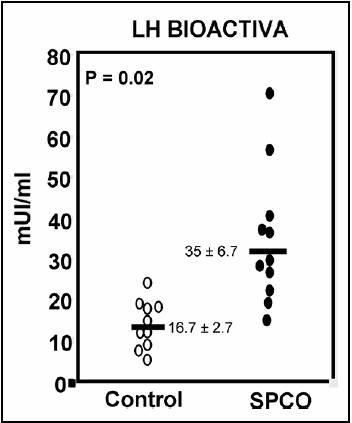
Figura 2: LH biológicamente activa determinada por ensayo de células intersticiales de rata (RICT) en adolescentes con SPCO y controles eumenorreicas. La línea horizontal representa la media para cada grupo.
Figura 3: Patrón de distribución de isoformas de LH por intervalo de pH en adolescentes normales y con SPCO. Las barras representan la proporción de LH recuperada en intervalos de 0.99 unidades de pH en relación al total de LH recuperada de cada cromatoenfoque. Cada barra representa la media ± DS de tres separaciones por cromatoenfoque independientes. * p = 0.031 vs. control en la misma banda de pH.
Algunos estudios disponibles en animales y una evaluación reciente en niños puberales demostraron que la estimulación por GnRH lleva a la producción por la glándula hipófisis de isoformas de LH más básicas(49,50). Las anormalidades neuroendocrinas en la secreción de LH que observamos en las adolescentes con SPCO pueden sugerir que una aceleración en la actividad del generador de pulsos de GnRH y/o una mayor capacidad de respuesta del gonadotropo podrían ser los responsables de la secreción predominante de isoformas básicas y más bioactivas en estas pacientes.
Diversos estudios en modelos animales han demostrado que los esteroides gonadales (andrógenos y estrógenos) pueden modificar la naturaleza bioquímica de las cadenas de oligosacáridos de las gonadotrofinas y por lo tanto modificar su biopotencia(51). Las evidencias en seres humanos acerca de los mecanismos de regulación por los esteroides gonadales, en particular sobre la composición de los restos hidrocarbonatos de LH, son escasas y menos directas(51). Se ha demostrado que existen variaciones en la bioactividad de LH durante el ciclo menstrual y que ésta aumenta en forma importante en la menopausia(52). En estudios clínicos también se comprobó que la infusión endovenosa de estradiol o de 5α-dihidrotestosterona y la administración oral de estrógenos, antiestrógenos no esteroideos o antiandrógenos modifican significativamente la bioactividad de LH en mujeres y hombres sanos(53). Sin embargo, no se conoce aún exactamente cómo los andrógenos y estrógenos controlan la biosíntesis, secreción, interconversión y/o la depuración de las diferentes isoformas de LH en los seres humanos. La heterogeneidad de carga de las moléculas de LH (medida en nuestro estudio por técnicas de cromatoenfoque), refleja variaciones en la estructura de los carbohidratos con diferente número de restos de ácido siálico o de residuos sulfato que le confieren a la molécula carga negativa(54). En adolescentes con SPCO, hemos observado que los niveles elevados de LH bioactiva y el mayor porcentaje de formas básicas se asocian con mayores niveles séricos de andrógenos (androstenodiona y testosterona) y su precursor (17-OH progesterona), por lo que es posible que la elevada relación andrógenos/estrógenos que caracteriza a este síndrome modifique la proporción y el tipo de moléculas de LH secretadas así como su biopotencia.
2. Secreción de FSH inmunoactiva normal y aumento de inhibina B.
Se ha demostrado que la concentración de FSH en mujeres adultas con el síndrome presenta una reducción de alrededor del 30 % comparadas con controles eumenorreicas en fase folicular temprana(33). Las adolescentes con SPCO que hemos estudiado no presentaron alteraciones de las características de secreción de FSH inmunoactiva ni en el grado de regularidad de la secreción (Ap En) (Tabla 3).
Tabla 3: Pulsatilidad de FSH en SPCO (utilizando IFMA y Cluster para el análisis de pulsos).
Se ha sugerido que un incremento en la cantidad de inhibina secretada por las células de la granulosa de los folículos antrales pequeños de los ovarios poliquísticos podría ser la causa de la secreción disminuida de FSH por el gonadotropo en el SPCO(55,56). Los primeros estudios utilizando el clásico RIA para la determinación de inhibina no observaban diferencias entre las pacientes SPCO y las controles(57). Sin embargo, estos resultados deben ser tomados con precaución dado que el RIA reconoce toda la familia de péptidos de las inhibinas en circulación (subunidad alfa libre y los dímeros biológicamente activos). Recientemente, dos grupos de investigadores utilizando ensayos inmunométricos para la determinación de inhibinas diméricas comunicaron resultados no coincidentes, tanto disminuidos(58) como elevados(59) en mujeres adultas con SPCO. Por otro lado, se ha demostrado que la secreción de inhibina B, forma de inhibina dimérica biológicamente activa que se produce preponderantemente en las primeras etapas de la fase folicular del ciclo menstrual, presenta una secreción de tipo episódica(59). En este mismo estudio se demostró que las mujeres adultas con SPCO secretan mayores cantidades de inhibina B y pierden las características de pulsatilidad regular que se observa en las controles normales en la fase folicular media(56).
En nuestra experiencia, los niveles en suero de inhibina B en adolescentes con SPCO (171 ± 24 pg/ml) fueron significativamente más altos que los observados en las adolescentes controles (91± 15 pg/ml, p<0.01, Figura 4). Los niveles del precursor pro-αC fueron similares entre ambos grupos (SPCO: 224 ± 84 pg/ml vs. control: 256 ± 69 pg/ml). Los niveles de Inhibina B correlacionaron en forma positiva y significativa con los niveles de androstenodiona sólo en el grupo SPCO (r = 0.55, p<0.05)(60). Estos hallazgos son similares a lo observado por Pigny y col. en un grupo de adultas con el síndrome(61). Los niveles de LH y FSH no correlacionaron con inhibina B en el grupo control ni en el grupo SPCO.
Figura 4: Panel A: Concentración en suero (media + ESM) de Inhibina B en el grupo SPCO y en el grupo control.
Panel B: Correlación entre niveles séricos de inhibina B y androstenodiona (A) en los grupos SPCO y control. La línea representa la regresión lineal sólo para el grupo SPCO.
Resulta difícil establecer si estos niveles elevados de inhibina B representan la iniciación de, o una respuesta a, la disfunción del eje hipotálamo - hipófiso - ovárico que caracteriza a este síndrome. El aumento de la secreción de LH en relación a la secreción de FSH también puede ser la causa de alteraciones en la formación de estos péptidos diméricos por parte de la granulosa en los folículos del ovario poliquístico. Se ha postulado que el exceso de LH en forma directa o indirectamente a través del incremento de andrógenos producidos en las células tecales podría provocar también un incremento en la síntesis de factores locales en las células tecales (como el factor de crecimiento transformante de tipo β, TGFβ) que actuando a nivel paracrino sobre las células de la granulosa induciría aumento de la dimerización de las subunidades libres de inhibina y consecuentemente en los niveles de inhibina B(62). Si el aumento de inhibina B que se observa en suero es reflejo del incremento de los niveles de este péptido en el microambiente folicular, es posible que este defecto contribuya a la persistencia de una cohorte de pequeños folículos con inadecuado desarrollo y perpetúe así la existencia de ciclos anovulatorios en estas pacientes.
La coexistencia de niveles elevados de inhibina B y niveles inmunoactivos normales de FSH no descarta la posibilidad de que la bioactividad y/o el tipo de isoformas de FSH secretadas se encuentren alteradas en adolescentes con SPCO.
Se han demostrado niveles normales de FSH biológicamente activa en fluido folicular de folículos provenientes de pacientes adultas con el síndrome(63). Hasta el momento no hemos determinado los niveles de FSH bioactiva ni el patrón de isoformas de FSH en el grupo de adolescentes con SPCO.
Posibles alteraciones en los mecanismos regulatorios hipofisarios e hipotalámicos
Como se ha detallado previamente el principal inhibidor de la frecuencia de pulsos de GnRH en los ciclos ovulatorios es el aumento de estradiol y progesterona en la mitad de la fase lútea.
Se ha demostrado claramente que las anormalidades bioquímicas en lo que respecta a la inadecuada secreción gonadotrófica en SPCO no pueden ser atribuidas a un defecto en el mecanismo de retroalimentación negativa de los estrógenos sobre la liberación de gonadotrofinas ya que la infusión de estrógenos durante 4 horas a mujeres con el síndrome provocó una rápida disminución de los niveles séricos de LH(64), con atenuación en la amplitud de los pulsos y disminución de la frecuencia de los mismos, similar a lo observado en mujeres normales. De la misma manera, la administración de citrato de clomifeno, que evalúa el mecanismo de feed back positivo del estradiol, produce un incremento cuanti- y cualitativo de los niveles de LH y FSH similar al observado en mujeres ciclantes que reciben este fármaco(64). Ambos datos indican la presencia de mecanismos intactos de retroalimentación negativa y positiva de estrógenos sobre la liberación de LH en pacientes con SPCO y sugieren la existencia de una sensibilización pituitaria probablemente como resultado de la exposición crónica a estrógenos acíclicos(65).
Las mujeres con SPCO son hiperrespondientes a la administración de GnRH. Esta hiperrespuesta que se manifiesta únicamente para la LH, es mayor en aquellas que presentan concentraciones basales más altas de esta hormona. Se ha sugerido que este aumento en la respuesta de LH sería secundario a un anormal mecanismo de retroalimentación pituitario por los niveles circulantes de andrógenos y/o estrógenos. Sin embargo, infundiendo estos esteroides a mujeres normales no se ha podido reproducir esta alteración(66).
Recientemente, se ha revelado una disminución en la sensibilidad del generador de pulsos de GnRH a la inhibición por progesterona(67). Esta anormalidad es revertida por el pretratamiento de estas pacientes con un antagonista del receptor de andrógenos como la flutamida, sugiriendo que los elevados niveles de andrógenos que persisten en el síndrome pueden alterar la sensibilidad hipotalámica del generador de pulsos de GnRH a la inhibición por los esteroides sexuales y llevar a un aumento de la secreción de LH(68). Así, las mujeres con SPCO, requerirían mayores niveles de progesterona para enlentecer al generador de pulsos de GnRH, resultando en una inadecuada síntesis de FSH y una persistente estimulación de LH sobre la secreción de andrógenos por el ovario(68)(70). Estos hallazgos apoyan, además, la hipótesis de la génesis del síndrome de poliquistosis ovárica durante la pubertad. En los estadios iniciales de la pubertad, el incremento de la secreción de LH asociado al sueño provoca un estímulo de la secreción de andrógenos por el ovario, los que subsecuentemente suprimen la frecuencia y amplitud de los pulsos de LH durante el día siguiente (generándose el ritmo diurno-nocturno de la secreción de LH). En niñas hiperandrogénicas que evolucionarán a SPCO, este incremento de esteroides nocturnos puede no ser adecuado para suprimir el generador de pulsos de GnRH, esto se traduciría en una secreción de LH con aumento de la frecuencia de pulsos de LH en forma persistente, secreción de FSH inadecuada y alteraciones en el programa de desarrollo folicular(69).
Además, uno de los primeros estudios en los que se evaluó el ritmo circadiano de LH utilizando RIA en un grupo pequeño de adolescentes con SPCO demostró por primera vez un patrón anormal de secreción de LH, caracterizado por una desincronización de la descarga de LH con el período de sueño nocturno(31). Las alteraciones en el ritmo circadiano de LH podrían indicar defectos primarios del generador de pulsos de GnRH en el desarrollo del síndrome, sobre todo teniendo en cuenta que esta anormalidad sólo se detectó en el patrón de secreción de LH siendo normales los perfiles secretorios de prolactina y cortisol en estas pacientes(31). Estudios más recientes realizados en adolescentes hiperandrogénicas confirmaron estos hallazgos y sugirieron que los mismos podrían indicar un patrón de evolución al desarrollo de SPCO(69). De cualquier modo el rol exacto de esta alteración cronobiológica en la patogénesis del SPCO se desconoce.
3. Alteración de la sincronía de secreción de LH-Andrógenos
La ausencia de ritmo circadiano de la secreción de LH y las anormalidades en los mecanismos de retrocontrol de los esteroides gonadales (andrógenos y progesterona) sobre la unidad GnRH-LH en pacientes con SPCO sugieren alteraciones de la sincronía de secreción de LH (hormona efectora) y andrógenos (hormonas subordinadas) en las pacientes con SPCO(70).
En nuestra experiencia, en adolescentes con SPCO, desde una perspectiva unihormonal (análisis de ApEn) los patrones de secreción de LH y androstenodiona (A) (aunque no la testosterona: T) fueron consistentemente más irregulares(70). Analizando la sincronía entre pares de hormonas relacionadas utilizando dos técnicas complementarias (cross ApEn y cross correlación), las pacientes con SPCO presentaron deterioro de las asociaciones entre LH-andrógenos comparadas con las adolescentes eumenorreicas. Las adolescentes eumenorreicas mostraron asociaciones significativas (sincronía de secreción) entre los pares de hormonas LH-T, LH-A y T-A (Figura 5, panel superior), en las pacientes con SPCO en cambio, el patrón de secreción de LH no correlacionó con el perfil de testosterona (Figura 5, panel inferior), mostrando una asociación débil sólo a los 0 min con el perfil de androstenodiona (Figura 5, panel inferior). Se verificó la interrelación entre los perfiles de andrógenos solamente en los tiempos 0 y -20 min (Figura 5, panel inferior). Estos hallazgos pueden indicar defectos en los mecanismos de control del eje gonadal en el síndrome de poliquistosis ovárica.

Figura 5: Cada gráfico representa los valores de la mediana y rango del coeficiente de cross correlación (r, eje Y) entre las concentraciones de pares de hormonas interdependientes en función de un barrido del tiempo de muestreo en minutos (lag) en 10 adolescentes control (panel superior) y 11 adolescentes con SPCO (panel inferior). Se aplicó el análisis de cross correlación a los perfiles espontáneos de LH - testosterona (T), LH - androstenodiona (A) y T - A. Los asteriscos representan el hallazgo de un coeficiente de correlación estadísticamente significativo a un determinado tiempo de retraso (lag).
Experimentos clínicos recientes indican que infusiones endovenosas de péptidos hipotalámicos (GnRH, GHRH, TRH y GH-RP-2) provocan una secreción más irregular de la hormona hipofisaria cuya secreción estimulan(71-73). Si estas observaciones pudieran generalizarse a ciertas situaciones fisiopatológicas podrían sugerir que un generador de pulsos de GnRH hipotalámico más autónomo (menos dependiente de los mecanismos de “feed back”) podría comandar e inducir un patrón de secreción de LH más irregular en el SPCO(74-76). Esta secreción de LH amplificada e irregular provocaría una alteración en la secreción de andrógenos ováricos.
La atenuación de los mecanismos de control que ejercen los andrógenos sobre la unidad GnRH/LH podría, adicionalmente, alterar la normal integración entre estos ejes.
Este escenario de tres defectos principales en el síndrome de poliquistosis ovárica (un generador de pulsos de GnRH más autónomo, una secreción de LH amplificada e irregular y un mecanismo de retrocontrol por andrógenos atenuado) puede a su vez ser modulado o mantenido por: 1) la producción de LH relativamente exacerbada por el hiperandrogenismo, hiperinsulinismo, hiperestrogenismo o factores intrahipofisarios específicos desconocidos(77) y 2) esteroidogénesis aumentada en las células tecales debido a defectos enzimáticos locales, amplificados por la hiperestimulación por LH o por insulina(78,79).
Sin duda, para probar estas hipótesis es necesario desarrollar modelos experimentales o ensayos clínicos que investiguen sobre estos defectos en los mecanismos de retroalimentación retrógrados y anterógrados en el eje hipotálamo-pituitario-gonadal.
Varios estudios han analizado el impacto del exceso de andrógenos sobre la actividad del generador de pulsos de GnRH. La administración por corto plazo de testosterona o de dihidrotestosterona a bajas dosis a mujeres adultas eumenorreicas no altera las concentraciones medias ni la amplitud de los pulsos de LH, mientras que altas dosis de testosterona disminuyen la frecuencia de pulsos y/o la secreción media de LH(80). Sin embargo, nuestro grupo ha demostrado recientemente que la infusión endovenosa de testosterona a bajas dosis (efecto a corto plazo) produce un efecto estimulatorio de la secreción de LH aumentando específicamente la altura de los pulsos y el nadir o valle de la secreción(81). El bloqueo del receptor de andrógenos con acetato de ciproterona(82), no disminuye la amplitud de los pulsos de LH, pero la flutamida normaliza las alteraciones en el retrocontrol negativo producida por progesterona exógena en mujeres con SPCO(83,84). De confirmarse este hallazgo implicaría que la hiperandrogenemia participa en la respuesta anormal del retrocontrol de la secreción de gonadotrofinas en mujeres con SPCO.
Nuestros hallazgos de deterioro en el orden de secreción uniglandular y desacople de la secreción bihormonal (LH-andrógenos) en adolescentes con SPCO son consistentes con defectos en el control de la secreción ovárica de andrógenos dependiente de LH y con una alteración en el control negativo que ejercen los andrógenos sobre la secreción GnRH-LH.
4. Hipersecreción de GH en adolescentes con SPCO con IMC normal
Las características del patrón de secreción de GH en SPCO han sido motivo de controversia. La mayoría de los estudios fueron realizados en mujeres adultas, utilizaron inmunoensayos de inadecuada sensibilidad y un tiempo de muestreo demasiado corto para evaluar la secreción espontánea de una hormona (Ej. 4 ó 6 horas)(85-88). Otros estudios no han tenido en cuenta la influencia negativa de la obesidad sobre la secreción de GH o no han explorado otros componentes del sistema de la hormona de crecimiento como los IGFs y sus proteínas transportadoras.
El primer estudio que utilizó una frecuencia de muestreo adecuada y ensayos apropiados para evaluar varios de los componentes del eje GHIGFs demostró un aumento de la amplitud de los pulsos de GH en mujeres adultas con IMC normal y SPCO en relación a las controles de similar IMC y a las obesas con el síndrome(37). En nuestra experiencia, combinando un ensayo ultrasensible para la determinación de GH (IFMA) y análisis de deconvolución para determinar las características de secreción y de depuración de esta hormona, el grupo de adolescentes con SPCO con IMC normal presentó mayor secreción de GH integrada en 12 hs. nocturnas y mayor tasa de secreción pulsátil de GH aunque la frecuencia de eventos secretorios fue normal en estas pacientes (Figura 6). Estos hallazgos plantean la hipótesis que en este síndrome, excluyendo el efecto negativo de la obesidad, la mayor secreción de GH está determinada por una modulación de la amplitud del pulso de GH.
Figura 6: Valores individuales y media de los distintos parámetros de la secreción de GH en adolescentes con SPCO y controles con IMC ≤ 25.
Se ha demostrado que los esteroides sexuales, estradiol (en forma directa) y testosterona (luego de la aromatización a estradiol), son los principales factores reguladores de la amplitud de los pulsos de GH(89-92). El incremento en la cantidad de andrógenos, por sí mismos o luego de la aromatización a estrógenos, podría modificar la cantidad y el patrón de secreción de GH en el SPCO.
Los esteroides pueden modificar la producción hipofisaria de GH modulando el perfil hipotalámico de las principales neurohormonas reguladoras de GH, es decir GHRH y somatostatina(93,94). Dado que la interacción entre estas dos neurohormonas determina el grado de regularidad de la secreción de GH, un disturbio en el microambiente esteroideo puede determinar cambios en el patrón de liberación de GH(95). Es bien conocido el dimorfismo sexual en el patrón de secreción de GH en roedores y menos marcado pero también evidente en humanos. Las hembras presentan un patrón de secreción de GH más irregular (mayores valores de ApEn) que los machos(96) probablemente por pérdida del efecto inhibitorio del mecanismo ultracorto de control de GH sobre el somatrotropo(97) y mayor liberación de somatostatina hipotalámica en el macho(98,99). En nuestro grupo de adolescentes con SPCO hemos observado que el patrón de liberación de GH fue más regular (menores valores de ApEn, una característica que se observa en el varón) comparadas con las controles eumenorreicas (Figura 6). Este hallazgo sugiere que el microambiente enriquecido de andrógenos que opera sobre la unidad hipotálamo hipofisaria podría provocar alteraciones en los mecanismos de control dependientes de GHRH y somatostatina modificando el perfil espontáneo de secreción de GH hacia un patrón masculino.
Se ha hipotetizado que el estado de hiposomatotropinismo que presentan las pacientes adultas obesas con SPCO está relacionado con modificaciones en la concentración de ghrelina. Se ha demostrado recientemente en pacientes con SPCO y resistencia a la insulina una franca disminución en los niveles séricos de ghrelina y que el tratamiento con metformina provocó un incremento de los niveles de este péptido(100). Lamentablemente en este estudio no se ha evaluado la secreción de GH antes y luego del tratamiento(100).
Los niveles séricos de IGF-1 total y de IGFBP-3 son normales en pacientes con SPCO(101). El IGF-1 también circula unido a IGFBP-1 (aunque en menor proporción que el que está unido a IGFBP-3). Se ha demostrado en pacientes con SPCO una franca disminución de los niveles de IGBP1 (probablemente como consecuencia del incremento en los niveles de insulina), lo que provoca un aumento de la relación molar de IGF-1/IGFBP-1(102). Therry van Dessel y col.(103) demostraron un aumento de la concentración de IGF-1 libre con niveles normales de IGF-1 total, IGF-2 e IGFBP-3 en adultas con SPCO.
Los estudios sobre la función hipotálamo hipofisaria que relacionen el eje gonadal y el de la hormona de crecimiento en adolescentes no obesas con SPCO son muy escasos. Recientemente, Ibáñez y col. han demostrado un incremento de la media de secreción y de la amplitud de los pulsos de GH en un grupo reducido de 8 adolescentes con hiperandrogenismo ovárico e hiperinsulinemia con antecedentes de bajo peso de nacimiento y pubarca precoz(104). En este mismo trabajo se demostró que la hipersecreción de GH se normalizó luego del tratamiento combinado con flutamida (antiandrógeno) y metformina (agente sensibilizador de la acción de la insulina). Esta terapia combinada provocó disminución de los andrógenos séricos, del grado de hirsutismo, aumento de la sensibilidad a la insulina y un perfil lipídico menos aterogénico así como una marcada redistribución de la masa grasa y de la masa magra resultando en una composición corporal más femenina y mejoró la tasa ovulatoria en estas pacientes(104). Si bien estos efectos beneficiosos son atribuibles principalmente a una mayor sensibilidad a la insulina y menor producción de andrógenos inducidas por el tratamiento, la disminución de la secreción de GH y de IGF-1 durante el tratamiento podrían ser también factores relevantes en la mejoría de la función ovárica.
Conclusiones
En relación con lo expuesto, proponemos que en adolescentes con SPCO el incremento en la actividad del generador de pulsos de GnRH en un microambiente hipofisario en el que predominan los andrógenos sobre los estrógenos podría promover la elevada secreción de LH debido a tres alteraciones neuroendocrinas principales: mayor tasa de secreción basal, elevada frecuencia y mayor masa de LH secretada por pulso. Además, del aumento en la cantidad de LH liberada en adolescentes con SPCO, el hallazgo de una secreción de LH más desordenada e irregular podría indicar anormalidades intrínsecas hipofisarias en el proceso de liberación de LH o reflejar alteraciones en los mecanismos de control por los andrógenos (o estrógenos o GnRH). Este estado de hipersecreción de LH se acompaña de un predominio de isoformas básicas y más bioactivas en las etapas iniciales del síndrome de poliquistosis ovárica.
El desacople de la secreción bihormonal (LH-A y LH-T) en adolescentes con SPCO es consistente con defectos en el control de la secreción ovárica de andrógenos dependiente de LH y con una alteración en el control negativo que ejercen los andrógenos sobre la secreción GnRH/LH. Estas alteraciones neuroendocrinas en la unidad GnRH/ LH y andrógenos ováricos podrían promover por sí mismas o en conjunción con otras hormonas (GH, leptina, insulina), y/o factores intraováricos locales (inhibinas, IGFs y BPs) el incremento de la secreción de andrógenos ováricos en este síndrome y consecuentemente inducir alteraciones en el desarrollo folicular y anovulación crónica.
Agradecimientos
La autora desea expresar su agradecimiento a los que en equipo han aunado esfuerzos para el estudio de las alteraciones neuroendocrinas del SPCO: Dra. María Cecilia García Rudaz, Dra. María Eugenia Escobar, Dra. Marta Barontini y Dr. Johannes Veldhuis. Además, quiere resaltar la colaboración de todos aquellos que aportaron su empeño y capacidad para la realización de los estudios hormonales: Téc. Ana María Montese, Téc. Mónica Campos, Téc. Perla Rossano y al personal de enfermería de la División de Endocrinología del Hospital de Niños Dr. Ricardo Gutiérrez.
1. Franks, S. Polycystic Ovary Syndrome. N Engl J Med 333: 853-861, 1995. [ Links ]
2. Yen S.S.C. Polycystic ovary syndrome (Hyperandrogenic chronic anovulation). En Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management (4th Edition). Yen S.S.C., Jaffe R.B., Barbieri R.L. WB Saunders Company, Philadelphia, USA, pag. 436-478, 1999. [ Links ]
3. Marshall J. C., Eagleson C. A. Neuroendocrine aspects of polycystic ovary syndrome. Endocrinol Metab Clin North Am 28: 295-324, 1999. [ Links ]
4. Sherman A. I.; Brown S. The precursors of endometrial carcinoma. Am J Obstet Gynecol 135: 947-956, 1979. [ Links ]
5. Wild R. A., Painter P. C., Coulson P. B. y col. Lipoprotein lipid concentrations and cardiovascular risk in women with polycystic ovary syndrome. J Clin Endocrinol Metab 61: 946-951, 1985. [ Links ]
6. Stein, I. F., Leventhal, M. L. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol 29: 181-191, 1935. [ Links ]
7. Stein, IF. Duration of fertility following ovarian wedge resection-stein-leventhal syndrome. West J Surg Obstet Gynecol 72: 237-242, 1964. [ Links ]
8. Ehrmann, D. A. Polycystic ovary syndrome. N Engl J Med 352: 1223-1236, 2005. [ Links ]
9. Zawadzki J. K., Dunaif A. Diagnostic criteria for polycystic ovary syndrome: Towards a rational approach. En Polycystic Ovary Syndrome. Dunaif, A.; Givens, J.R.; Haseltine, F.P.; Merriam, G.R. Blackwell Scientific, Boston, USA, pag. 377, 1992. [ Links ]
10. Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 81: 19-25, 2004. [ Links ]
11. Rotterdam ESHRE/ASRM-Sponsored PCOS consensus workshop group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 19: 41-47, 2004. [ Links ]
12. Azziz, R. Diagnosis of polycystic ovarian syndrome: the Rotterdam criteria are premature. J Clin Endocrinol Metab 91: 781-785, 2006. [ Links ]
13. Franks, S. Diagnosis of polycystic ovarian syndrome: in defense of the Rotterdam criteria. J Clin Endocrinol Metab 91: 786-789, 2006. [ Links ]
14. Azziz R., Carmina, E., Dewailly D. y col. Position statement: Criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society Guidline. J Clin Endocrinol Metab 91: 4237-4245, 2006. [ Links ]
15. Rosenfield R. L. Plasma free androgen patterns in hirsute women and their diagnostic implications. Am J Med 66: 417, 1979. [ Links ]
16. Polson D., Adams J., Wadsworth J. y col. Polycystic ovaries-a common finding in normal women. Lancet 1: 870, 1988. [ Links ]
17. Taieb J., Mathian B., Millot F. y col. Testosterone measured by 10 immunoassays and by isotope-dilution gas chromatography-mass spectrometry in sera from 116 men, women, and children. Clin Chem 49: 1381, 2003. [ Links ]
18. Clayton R. N., Ogden V., Hodgkinson J. y col. How common are polycystic ovaries in normal and what is their significance for the fertility of the population? Clin Endocrinol (Oxf) 37: 127-134, 1992. [ Links ]
19. Rosenfield R. L., Ghai K, Ehrmann D. A. y col. Diagnosis of polycystic ovary syndrome in adolescence. Comparison of adolescent and adult hyperandrogenism. J Pediatr Endocrinol Metab 13: 1285, 2000. [ Links ]
20. Buggs C., Rosenfield R. L. Polycystic ovary syndrome in adolescence. Endocrinol Metab Clin N Am 34: 677-705, 2005. [ Links ]
21. Venturoli S., Porcu E., Fabbri R. y col. Longitudinal change of sonographic ovarian aspects and endocrine parameters in irregular cycles of adolescence. Pediatr Res 38: 974, 1995. [ Links ]
22. Adams J., Franks S., Polson D. W. y col. Multifollicular ovaries: clinical and endocrine features and response to pulsatile gonadotropin releasing hormone. Lancet 2: 1375-1379, 1985. [ Links ]
23. Belosi C., Giuliani M., Suriano R. y col. Diagnosis of polycystic ovary syndrome. Minerva Ginecol 56: 7-13, 2004. [ Links ]
24. Franks S. Adult polycystic ovary syndrome begins in childhood. Best Pract Res Clin Endocrinol Metab 16: 263-272, 2002. [ Links ]
25. Ibanez L., Valls C., Potau N. y col. Polycystic ovary syndrome after precocious pubarche: ontogeny of the low-birthweight effect. Clin Endocrinol (Oxf) 55: 667-672, 2001. [ Links ]
26. Abbott D. H., Dumesic D. A., Franks S. Developmental origin of polycystic ovary syndromea hypothesis. J Endocrinol 174: 1-5, 2002. [ Links ]
27. Escobar M. E., Ropelato M. G., Ballerini M. G. y col. Acceleration of LH pulse frequency in adolescent girls with a history of central precocious puberty with versus without hyperandrogenism. Horm Res (en prensa), 2007. [ Links ]
28. Venturoli S., Porcu E., Fabbri R. y col. Post-menarchal evolution of endocrine pattern and ovarian aspects in adolescents with menstrual irregularities. Fertil Steril 48: 78-85, 1987. [ Links ]
29. Apter D., Butzow T. L., Laughlin G. A., y col. Gonadotropin-releasing hormone pulse generator activity during pubertal transition in girls: pulsatile and diurnal patterns of circulating gonadotropins. J Clin Endocrinol Metab 76: 940-949, 1993. [ Links ]
30. Siegberg R., Nilsson C. G., Stenman U. H. y col. Endocrinologic features of oligomenorrheic adolescent girls. Fertil Steril 46: 852-857, 1986. [ Links ]
31. Zumoff B., Freeman R., Coupey S. y col. A chronobiologic abnormality in luteinizing hormone secretion in teenage girls with the polycystic-ovary syndrome. N Engl J Med 309: 1206-1209, 1983. [ Links ]
32. Apter D., Butzow T., Laughlin G. A., y col. Accelerated 24-hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: Relevance to the developmental phase of polycystic ovarian syndrome. J Clin Endocrinol Metab 79: 119-125, 1994. [ Links ]
33. Yen S. S. C., Vela P., Rankin J. Inappropriate secretion of follicle-stimulating hormone and luteinizing hormone in polycystic ovarian disease. J Clin Endocrinol Metab 30: 435-442, 1970. [ Links ]
34. Rebar R., Judd H. L., Yen S. S. C. y col. Charaterization of the inappropriate gonadotropin secretion in polycystic ovary syndrome. J Clin Invest 57: 1320-1329, 1976. [ Links ]
35. Leondires M. P., Berga S.L. Role of GnRH drive in the pathophysiology of polycystic ovary syndrome. J Endocrinol Invest 21: 476-485, 1998. [ Links ]
36. García Rudaz M. C., Ropelato M. G., Veldhuis J. D. y col. Augmented frequency and mass of LH discharged per burst are accompanied by marked disorderliness of LH secretion in adolescents with polycystic ovary syndrome (PCOS). Eur J Endocrinol 139: 621-630; 1998. [ Links ]
37. Morales A. J., Laughlin G. A., Butzow T. y col. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: Common and distinct features. J Clin Endocrinol Metab 81: 2854-2864, 1996. [ Links ]
38. Taylor A. E., McCourt B., Martin K. A. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab 82: 2248-2256; 1997. [ Links ]
39. Arroyo A., Laughlin G. A., Morales A. J. Inappropriate gonadotropin secretion in polycystic ovary syndrome: Influence of adiposity. J Clin Endocrinol Metab 82: 3728-3733, 1997. [ Links ]
40. Anttila, L.; Ding, Y.Q.; Ruutiainen, K. Clinical features and circulating gonadotropin, insulin, and androgen interactions in women with polycystic ovarian disease. Fertil Steril 55: 1057-1061, 1991. [ Links ]
41. Mantzoros C. S. Role of leptin in reproduction. Ann N Y Acad Sci 900: 174-183, 2000. [ Links ]
42. Brzechffa P. R., Jakimiuk A. J., Agarwal S. K. Serum immunoreactive leptin concentrations in women with polycystic ovary syndrome. J Clin Endocrinol Metab 81: 4166-4169, 1996. [ Links ]
43. Mantzoros C. S., Dunaif A., Flier J. S. Leptin concentrations in the polycystic ovary syndrome. J Clin Endocrinol Metab 82: 1687-1691, 1997. [ Links ]
44. Laughlin G. A., Morales A. J., Yen, S. S. C. Serum leptin levels in women with polycystic ovary syndrome: The role of insulin resistance/hyperinsulinemia. J Clin Endocrinol Metab 82: 1692-1696, 1997. [ Links ]
45. Licinio J., Negrao A. B., Mantzoros C. y col. Synchronicity of frequently sampled, 24-h concentrations of circulating leptin, luteinizing hormone, and estradiol in healthy women. Proc Natl Acad Sci USA 95: 2541-2546, 1998. [ Links ]
46. Ropelato, M.G.; García Rudaz, M.C.; Castro- Fernández, C. y col. A preponderance of basic luteinizing hormone (LH) isoforms accompanies inappropriate hypersecretion of both basal and pulsatile LH in adolescents with polycystic ovarian syndrome. J Clin Endocrinol Metab 84: 4629-4636; 1999. [ Links ]
47. Ding, Y. Q.,s Huhtaniemi, I. Preponderance of basic isoforms of serum luteinizing hormone (LH) is associated with the high bio/immuno ratio of LH in healthy women and in women with polycystic ovarian disease. Hum Reprod 6: 346-350, 1991. [ Links ]
48. Imse V., Holzapfel G., Hinney B., y col. Comparison of luteinizing hormone pulsatility in the serum of women suffering from polycystic ovarian disease using a bioassay and five different immunoassays. J Clin Endocrinol Metab 74: 1053-1061, 1992. [ Links ]
49. Baldwin D. M., Highsmith R. F. Ramey J. W. An in vitro study of LH release, synthesis and heterogeneity in pituitaries from proestrous and short-term ovariectomized rats. Biol Reprod 34: 304-315, 1986. [ Links ]
50. Phillips D. J., Wide L. Serum gonadotropin isoforms become more basic after an exogenous challenge of gonadotropin-releasing hormone in children undergoing pubertal development. J Clin Endocrinol Metab 79: 814-819,1994. [ Links ]
51. Dufau M. L., Veldhuis J. D. Pathophysiological relationships between the biological and immunological activities of luteinizing hormone. Baillieres Clin Endocrinol Metab 1: 153-176; 1987. [ Links ]
52. Veldhuis J. D., Beitins I. Z., Johnson M. L. y col. Biologically active luteinizing hormone is secreted in episodic pulsations that vary in relation to stage of the menstrual cycle. J Clin Endocrinol Metab 58: 1050-1058, 1984. [ Links ]
53. Veldhuis J. D., Dufau M. L. Steroidal regulation of biologically active luteinizing hormone secretion in men and women. Hum Reprod 8:(Suppl 2): 84-96,1993. [ Links ]
54. Baenziger J. U., Green E. D. Pituitary glycoprotein hormone oligosaccharides: Structure, synthesis and function of the asparaginelinked oligosaccharides on lutropin, follitropin and thyrotropin. Biochim Biophys Acta 947: 287-306, 1988. [ Links ]
55. Hurkadli K. S., Jayaraman S., Gopalakrishnan K. y col. Role of inhibin in polycystic ovarian syndrome. Int J Fertil 31: 165-169,1986. [ Links ]
56. Lockwood G. M., Muttukrishna S., Groome N. P., y col. Mid-follicular phase pulses of inhibin B are absent in polycystic ovarian syndrome and are initiated by successful laparoscopic ovarian diathermy: A possible mechanism regulating emergence of the dominant follicle. J Clin Endocrinol Metab 83: 1730-1735, 1998. [ Links ]
57. Buckler H. M., McLachlan R. I., McLachlan V. B. y col. Serum inhibin levels in polycystic ovary syndrome: Basal levels and response to luteinizing hormone-releasing hormone agonist and exogenous gonadotropin administration. J Clin Endocrinol Metab 66: 798-803; 1988. [ Links ]
58. Lambert-Messerlian G. M., Hall J. E. Sluss, P. M. y col. Relatively low levels of dimeric inhibin circulate in men and women with polycystic ovarian syndrome using a specific two-site enzyme-linked immunosorbent assay. J Clin Endocrinol Metab 79: 45-50, 1994. [ Links ]
59. Mizunuma H., Andoh K., Obara, M. y col. Serum immunoreactive inhibin levels in polycystic ovarian disease (PCOD) and hypogonadotropic amenorrhea. Endocr J 41: 409-414, 1994. [ Links ]
60. Ropelato M. G., García-Rudaz M. C., Trigo R.V. y col. Inhibina B sérica en adolescents con poliquistosis ovárica. Libro de Resúmenes de la XV Reunión Anual Sociedad Latinoamericana de Endocrinología Pediátrica. Punta del Este, 27-31 de Octubre de 2002. Uruguay. [ Links ]
61. Pigny P., Desailloud R., Cortet-Rudelli C., y col. Serum alpha-inhibin levels in polycystic ovary syndrome: Relationship to the serum androstenedione level. J Clin Endocrinol Metab 82: 1939-1943, 1997. [ Links ]
62. Lanuza G. M., Groome N. P., Barañao J. L. y col. Dimeric inhibin A and B production care differentially regulated by hormones and local factors in rat granulose cells. Endocrinology 140: 2549-2554, 1999. [ Links ]
63. Erickson G. F., Magoffin D. A., Garzo V. G. y col. Granulosa cells of polycystic ovaries: Are they normal or abnormal?. Hum Reprod 7: 293-299, 1992. [ Links ]
64. Tsai C. C., Yen, S. S. C. Acute effects of intravenous infusion of 17-beta-estradiol on gonadotropin release in pre- and post-menopausal women. J Clin Endocrinol Metab 32: 766-771, 1971. [ Links ]
65. Baird D. T., Corker C. S., Davidson D. W. y col. Pituitary-ovarian relationships in polycystic ovary syndrome. J Clin Endocrinol Metab 45: 798-801, 1977. [ Links ]
66. Chang R. J., Mandel F. P. Lu J. K. y col. Enhanced disparity of gonadotropin secretion of estrone in women with polycycstic ovarian disease. J Clin Endocrinol Metab 54: 490-494; 1982. [ Links ]
67. Pastor C. L., Griffin-Korf M. L. Aloi J. A. y col.Polycystic ovary syndrome: Evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab 83: 582-590, 1998. [ Links ]
68. McCartney, C.R.; Eagleson, C.A.; Marshall, J.C. Regulation of gonadotropin secretion: Implications for polycystic ovary syndrome. Semin Reprod Med 20: 317-326, 2002. [ Links ]
69. Porcu E., Venturoli S., Magrini O. y col. Circadian variations of luteinizing hormone can have two different profiles in adolescent anovulation. J Clin Endocrinol Metab 65: 488-493, 1987. [ Links ]
70. Veldhuis J. D.; Pincus S. M. García-Rudaz, M. C. y col. Disruption of the joint synchrony of luteinizing hormone, testosterone, and androstenedione secretion in adolescents with polycystic ovarian syndrome. J Clin Endocrinol Metab 86: 72-79. 2001. [ Links ]
71. Veldhuis J. D., Wilkowski M. J., Zwart A. D. y col. Evidence for attenuation of hypothalamic gonadotropin-releasing hormone (GnRH) impulse strength with preservation of GnRH pulse frequency in men with chronic renal failure.J Clin Endocrinol Metab76: 648-654, 1993. [ Links ]
72. Iranmanesh A., South S., Liem A. Y., y col. Unequal impact of age, percentage body fat, and serum testosterone concentrations on the somatotropic, IGF-I, and IGF-binding protein responses to a three-day intravenous growth-hormone- releasing-hormone (GHRH) pulsatile infusion. Eur J Endocrinol. 139: 59-71, 1998. [ Links ]
73. Shah N., Evans W. S., Bowers C. Y. y col. Tripartite neuroendocrine activation of the human growth-hormone (GH) axis in women by continuous 24-hour GH-releasing peptide (GHRP-2) infusion: pulsatile, entropic, and nyctohemeral mechanisms. J Clin Endocrinol Metab 84: 2140-2150, 1999. [ Links ]
74. Kazer R. R., Kessel B., Yen S.S.C. Circulating luteinizing hormone pulse frequency in women with polycystic ovary syndrome. J Clin Endocrinol Metab 65: 233-236, 1987. [ Links ]
75. Waldstreicher J., Santoro N. F., Hall J. E. y col. Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: Indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab 66: 165-172, 1988. [ Links ]
76. Berga S. L., Guzick D. S., Winters S. J. Increased luteinizing hormone and alpha- subunit secretion in women with hyperandrogenic anovulation. J Clin Endocrinol Metab 77: 895-901, 1993. [ Links ]
77. Adashi E. Y., Hsueh A. J. W., Yen S. S. C. Insulin enhancement of luteinizing hormone and follicle stimulating hormone release by cultured pituitary cells. Endocrinology 108: 1441-1449, 1981. [ Links ]
78. Barbieri R. L., Makris A., Ryan K. J. Insulin stimulates androgen accumulation in incubations of human ovarian stroma and theca. Obstet Gynecol 64: 73S-80S, 1984. [ Links ]
79. Fulghesu A. M., Villa P., Pavone V. y col. The impact of insulin secretion on the ovarian response to exogenous gonadotropins in polycystic ovary syndrome. J Clin Endocrinol Metab 82: 644-648, 1997. [ Links ]
80. Dunaif, A. Do androgens directly regulate gonadotropin secretion in the polycystic ovary syndrome?. J Clin Endocrinol Metab 63: 215-221, 1986. [ Links ]
81. Ropelato M. G., García Rudaz M. C., Escobar M. E. y col. Efecto de la infusión de testosterona (T) sobre la secreción de LH en adolescentes eumenorreicas y adolescentes con síndrome de poliquistosis ovárica (PCO). Libro de Resúmenes de la III Jornada Científica del Consejo de Investigación en Salud. (Abstract) pag: 8-9, 2005. [ Links ]
82. Couzinet B., Le Strat N., Brailly S. y col. Comparative effects of cyproterone acetate or a long-acting gonadotropin-releasing hormone agonist in polycystic ovarian disease. J Clin Endocrinol Metab 63: 1031-1035, 1986. [ Links ]
83. Eagleson C. A., Gingrich M. B., Pastor C. L. y col. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin- releasing hormone pulse generator to inhibition by estradiol and progesterone.J Clin Endocrinol Metab 85: 4047-4052, 2000. [ Links ]
84. Adams J. M., Taylor A. E., Crowley W. F. Jr. y col. Polycystic ovarian morphology with regular ovulatory cycles: insights into the pathophysiology of polycystic ovarian syndrome.J Clin Endocrinol Metab 89: 4343-4350, 2004. [ Links ]
85. Kazer R. R., Unterman T. G., Glick R. P. An abnormality of the growth hormone/insulinlike growth factor-I axis in women with polycystic ovary syndrome. J Clin Endocrinol Metab 71: 958-962, 1990. [ Links ]
86. Slowinska-Srzednicka J., Zgliczynski W., Makowska A. y col. An abnormality of the growth hormone/insulin-like growth factor- I axis in women with polycystic ovary syndrome due to coexistent obesity. J Clin Endocrinol Metab 74: 1432-1435, 1992. [ Links ]
87. Kaltsas T., Pontikides N., Krassas G. E. y col. Effect of gonadotrophin-releasing hormone agonist treatment on growth hormone secretion in women with polycystic ovarian syndrome. Hum Reprod 13: 22-26, 1998. [ Links ]
88. Prelevic G. M., Wurzburger M. I., Balint- Peric L. y col. Twenty-four-hour serum growth hormone, insulin, C-peptide and blood glucose profiles and serum insulin-like growth factor-I concentrations in women with polycystic ovaries. Horm Res 37:125-131, 1992. [ Links ]
89. Mauras N., Rogol A. D., Veldhuis, J. D. Increased hGH production rate after low-dose estrogen therapy in prepubertal girls with Turner's syndrome. Pediatr Res 28:626-630, 1990. [ Links ]
90. Weissberger A. J., Ho K. K. Activation of the somatotropic axis by testosterone in adult males: Evidence for the role of aromatization. J Clin Endocrinol Metab 76: 1407-1412, 1993. [ Links ]
91. Giustina A., Scalvini T., Tassi C. y col. Maturation of the regulation of growth hormone secretion in young males with hypogonadotropic hypogonadism pharmacologically exposed to progressive increments in serum testosterone. J Clin Endocrinol Metab 82: 1210-1219, 1997. [ Links ]
92. Veldhuis J. D., Zwart A. D., Iranmanesh A. Neuroendocrine mechanisms by which selective Leydig cell castration unleashes increased pulsatile LH release. Am J Physiol 272: R464-R474, 1997. [ Links ]
93. Iranmanesh A., Lizarralde G., Veldhuis J. D. Age and relative adiposity are specific negative determinants of the frequency and amplitude of growth hormone (GH) secretory bursts and the half-life of endogenous GH in healthy men. J Clin Endocrinol Metab 73: 1081-1088, 1991. [ Links ]
94. Devesa J., Lois N., Arce V. y col. The role of sexual steroids in the modulation of growth hormona (GH) secretion in humans. J Steroid Biochem Mol Biol 40:165-173, 1991. [ Links ]
95. Wehrenberg W. B., Giustina A. Basic counterpoint: Mechanisms and pathways of gonadal steroid modulation of growth hormone secretion. Endocr Rev 13: 299-308, 1992. [ Links ]
96. Veldhuis J. D., Metzger D. L., Martha P. M. Jr. y col. Estrogen and testosterone, but not a nonaromatizable androgen, direct network integration of the hypothalamo-somatotrope (growth hormone)-Insulin-like growth factor I axis in the human: Evidence from pubertal pathophysiology and sex-steroid hormone replacement. J Clin Endocrinol Metab 82: 3414-3420, 1997. [ Links ]
97. Pincus S. M., Gevers E. F., Robinson I. C. y col. Females secrete growth hormone with more process irregularity than males in both humans and rats. Am J Physiol 270: E107-E115, 1996. [ Links ]
98. Clark R. G., Robinson I. C. Growth hormone responses to multiple injections of a fragment of human growth hormone-releasing factor in conscious male and female rats. J Endocrinol 106: 281-289,1985. [ Links ]
99. Plotsky P. M., Vale W. Patterns of growth hormone-releasing factor and somatostatin secretion into the hypophysial-portal circulation of the rat. Science 25(230): 461-463, 1985. [ Links ]
100. Schofl C., Horn R., Schill T. y col. Circulating ghrelin levels in patients with polycystic ovary syndrome. J Clin Endocrinol Metab 87: 4607-4610, 2002. [ Links ]
101. Giustina A., Veldhuis J. D. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 19: 717-797, 1998. [ Links ]
102. Garcia-Rudaz M. C., Ropelato M. G., Escobar ME, Veldhuis J. D. y col. Amplified and orderly growth hormone (GH) secretion characterizes lean adolescents with polycystic ovary syndrome. Eur J Endocrinol. 147: 207-216, 2002. [ Links ]
103. Thierry van Dessel H. J., Lee P. D., Faessen G. y col. Elevated serum levels of free insulinlike growth factor I in polycystic ovary syndrome. J Clin Endocrinol Metab 84: 3030-3035, 1999. [ Links ]
104. Ibanez L., Ong K., Ferrer A. y col. Low-dose flutamide-metformin therapy reverses insulin resistance and reduces fat mass in nonobese adolescents with ovarian hyperandrogenism. J Clin Endocrinol Metab 88: 2600-2606, 2003. [ Links ]

