










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de endocrinología y metabolismo
versión On-line ISSN 1851-3034
Rev. argent. endocrinol. metab. vol.47 no.3 Ciudad Autónoma de Buenos Aires jul./set. 2010
REVISIÓN
Growth Hormone Deficiency (GHD) in Adults: To Treat or Not To Treat?
Deficiencia de hormona de crecimiento en el adulto ¿debe tratarse?
Cesar Luiz Boguszewski, MD, PhD
Correspondence to: Cesar Luiz Boguszewski. Associate Professor of Endocrinology, Department of Internal Medicine, SEMPR, Federal University of Parana, Curitiba, Brazil. Avenida Agostinho Leao Junior, 285. Curitiba, PR, Brazil – CEP 80030-110 Tel: +55 41 3360 7876 / Fax +55 41 3523 9184. cesarluiz@hc.ufpr.br
ABSTRACT
Over the last decade, different guidelines have been published for the diagnosis, treatment and monitoring of adult growth hormone deficiency (AGHD). Themes and recommendations common to the guidelines offer a pragmatic approach to the management of AGHD. Nevertheless, there is a need for more research in some key areas in which recommendations in the guidelines are supported by moderate evidence, at best. Recent meta-analysis and long-term follow-up studies have contributed with valuable information on the efficacy and safety of GH therapy in adults. This review brings a historical perspective of the AGHD, with an emphasis on the following aspects: (I) who are the appropriate candidates for GH therapy in adult life? (II) how to make the diagnosis (III) the impact of GH therapy; (IV) which therapeutic approach should be used? (V) how to follow and monitor the patients; and (VI) special aspects on mortality and longevity related to the GH-IGF-1 axis.
No financial conflicts of interest exist.
Key words: Growth hormone; Deficiency; Hypopituitarism; Young adult; Hormone replacement therapy.
INTRODUCTION
In 1962, Raben was the first to treat a 35-year-old hypopituitary woman with pituitary GH (1). After two months of treatment, the patient noted improvement in physical vigor and psychological well-being. However, due to the limited supply of pituitary GH at that time, the therapy was restricted to children with severe growth retardation until the introduction
of recombinant human GH in 1985. The abundance of biosynthetic GH radically changed its applications in clinical practice and opened several fascinating therapeutic possibilities.
Doubtless, one of the most important developments in the GH field was the establishment of a new entity called "syndrome of adult GH deficiency (AGHD)”(2). The definition and initial characterization of AGHD occurred in parallel with an epidemiologic study reporting increased mortality in patients with hypopituitarism, mainly related to cardiovascular diseases(3), which are confirmed by subsequent reports(4, 5). From these studies, the question was raised whether untreated AGHD could be responsible for this finding. However, even after twenty-five years, it is not possible to answer with certainty whether the increased mortality of hypopituitary patients is caused by AGHD per se or by other factors, such as previous exposure to radiation, the etiology of hypothalamic-pituitary disease or mistreatment of other associated pituitary hormonal deficiencies (TSH, LH / FSH and ACTH). Regardless of that, GH therapy in AGHD has been shown to improve many cardiovascular parameters related to increased morbidity and mortality in hypopituitarism(2, 6-8).
The AGHD should not be confused with the so-called "somatopause”, a physiological situation characterized by progressive decline of GH and IGF-1 production related to the aging process(9, 10). At this moment, neither the safety nor the benefits of GH administration to reestablishing 'youthful' GH levels in pituitary-replete adults have been defined to justify the prescription of GH in this situation(11-14). Thus, this review will focus on who are the appropriate adult GHD candidates for GH therapy, the correct biochemical criteria to define the diagnosis, and the main aspects of the replacement GH therapy in AGHD, mainly based on recent guidelines, meta-analysis and long-term studies. Finally, some aspects on mortality and longevity associated with the GH-IGF-1 axis will be briefly reviewed.
WHO ARE THE APPROPRIATE CANDIDATES FOR GH THERAPY?
There is broad consensus that all patients who show evidence of a disease affecting the hypothalamic-pituitary region, and in whom there is an intention to treat with GH, are appropriate candidates for testing for adult GHD (6-8). Accordingly, adult GHD is not synonymous with the "somatopause”, which refers to the aged-related physiological decline in GH and IGF-1 secretion. Advancing age is associated with a marked decrease in the 24-hour integrated GH concentrations, fraction of GH secreted in pulses, mean pulse amplitude and duration(9-10), but no study thus far has proved that GH therapy is beneficial and safe for otherwise healthy elderly patients(11-14). As a result, patients should not be tested for GHD based on the presumption of agerelated declines in GH alone. In addition, there is no indication to evaluate GH status and to replace GH in adult life in individuals with no evidence of hypothalamic-pituitary disease, functional GHD due to visceral obesity, and in short children treated with GH for other non-GHD pediatric indications (e.g., Turner syndrome, small for gestational age, idiopathic short stature) (Figure 1).
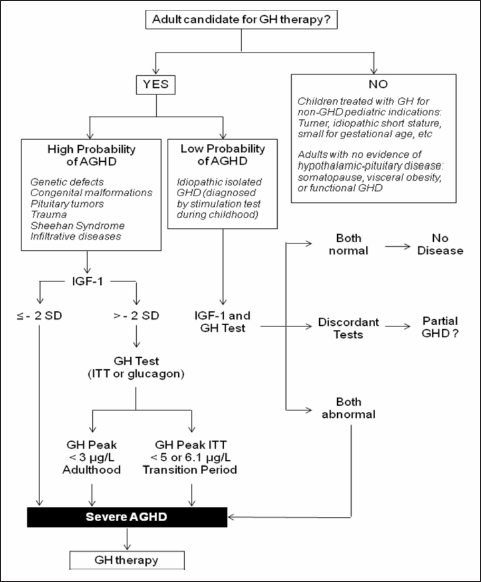
Figure 1. Algorithm for biochemical evaluation of adult growth hormone deficiency (AGHD). Glucagon test is presented as the alternative choice for ITT as GHRH production has been discontinued. SD (standard deviation) for IGF-1 levels according to reference values adjusted for age.
The relatively loose definition of the patients that might be appropriate candidates for GH treatment reflects the fact that AGHD is a heterogeneous entity that can arise from numerous etiologies (Figure 1). For example, genetic defects, congenital malformations, brain injury, sub-arachnoid haemorrhage (SAH), non-functioning and functioning pituitary adenomas (e.g. prolactinoma, Cushing's disease), craniopharyngiomas, infiltrative, inflammatory and vascular diseases, can all lead to AGHD. The condition can also arise as an iatrogenic consequence of the management of acromegaly and after neurosurgery and radiotherapy.
Many other aspects of the disease underscore the inherent heterogeneity. First, a group of patients presents isolated AGHD, while others suffer from multiple pituitary hormone deficiencies (combined AGHD). Second, idiopathic isolated GHD diagnosed by GH tests during childhood may resolve after puberty or persist into adulthood(15). In our hands, 56% of patients with this diagnosis during childhood had no biochemical criteria for AGHD when retested in adult life, while 94% of those with combined GHD during childhood persisted with AGHD(16). Third, in several cases the GHD begins de novo in adulthood. Finally, only patients with severe GHD (as established by strict biochemical criteria) are candidates for GH replacement therapy in adulthood, as partial GHD (or GH insufficiency) is still a poorly defined entity in which GH therapy is not indicated(17).
HOW TO MAKE THE DIAGNOSIS OF AGHD?
Clinical findings of adult GHD are nonspecific and not useful for diagnosis of AGHD, which depends on well-established biochemical criteria. Insulin tolerance test (ITT) and stimulation test using the combination of GHRH and arginine (GHRH-ARG) have been set as the diagnostic investigations of choice for AGHD(6-8). However, in July 2008, the only manufacturer of recombinant GHRH in the United States announced the discontinuation of its production, which raised the problem to consider another alternative GH stimulation test to replace the GHRH-ARG test. The GHRH plus GH-releasing peptide (GHRH-GHRP) test poses the same problem, making the glucagon test the best available alternative for cases where ITT is unsuitable or contraindicated(18). In Brazil, GHRH was never available in the clinical practice and the glucagon test has been used as an alternative to ITT for a long time, since the pioneer study done by Conceicao and co-workers from University of Río de Janeiro that, which showed a good performance and great diagnostic accuracy of the glucagon test for the diagnosis of AGHD(19). In contrast, provocative tests with clonidine, L-dopa and arginine can not be used in the diagnosis of AGHD(6-8). Each diagnostic test has advantages and limitations that clinicians need to consider. In addition, clinicians must take into account the substantial heterogeneity among GH assays and must be aware of which assay has been used for GH measurements. In general, an impaired peak GH level on one test is sufficient to diagnose AGHD in an appropriate setting(6-8).
Peak GH values are influenced by several factors, including age, adiposity and the type of GH secretagogue. For the ITT and glucagon test, the validated cutoff for biochemical diagnosis of severe AGHD is a peak GH response below 3 μg/l(6-8). However, a cutoff value of 5 μg/l(20) or 6.1 μg/l(21) in the ITT is recommended for the transition period – which has been arbitrarily defined as "a 6-7 years interval spanning from late puberty (after achievement of final height) to the full adult somatic maturation”(20).
Not all adult patients require a GH stimulation test for diagnosis of GHD. Patients with high probability of AGHD, as shown in Figure 1, including those with three or more pituitary deficiencies and low IGF-1 levels, do not require testing(6-8). In fact, low IGF-1 levels in the absence of catabolic conditions and liver disease are highly indicative of severe AGHD in patients with hypopituitarism, especially in those younger than 40 years and BMI below 25 kg/m2. Nevertheless, two points on IGF-1 measurements should be highlighted: (1) normal serum IGF-1 level does not exclude AGHD at any age; (2) there is still a need for normative age and gender data for IGF-1 levels in different clinical settings(6-8, 22). Current algorithm using GH tests and IGF-1 measurements for biochemical diagnostic approach is summarized in Figure 1, showing the differences between adult-onset GHD and the special criteria applied in the transition period.
IMPACT OF GH THERAPY ON AGHD
A growing body of evidence summarized in the guidelines shows that GH therapy results in clinical benefits for GHD patients at different ages(23). In the transition period, GH treatment should be continued in those young adults with persistent GHD after attaining final height to achieve full somatic development of bone and muscles. GH treatment in AGHD improves body composition, exercise capacity, skeletal integrity and quality of life (QoL)(6-8). A meta-analysis of placebo-controlled trials showed an improvement on cardiovascular risk markers, including diastolic blood pressure, total cholesterol and LDL-cholesterol(24). Similarly, other recent meta-analysis found strong evidence that GH replacement improves exercise performance in AGHD(25). GH therapy is more likely to benefit those patients who have more severe clinical and biochemical GHD compared with those with a less marked disease(6-8). In fact, there have been controversies in the therapeutic indications for GH(26, 27), especially related to those patients with no or minimal impairments(28). An important aspect to emphasize is the need to optimize replacement therapies for other pituitary deficiencies before starting GH. For instance, the avoidance of cortisol over-replacement is fundamental to improve some morbidities in patients with hypopituitarism(29).
Physicians should expect a large intra-individual variation in the clinical response to GH therapy in AGHD, which is mainly dependent on gender, age, BMI, GH dose, and the route of estrogen replacement in hypopituitary women(30, 31). In a collaborative study with the University of Gothemburg, Sweden, we have recently developed mathematical models based on clinical parameters for predicting changes in serum IGF-1 and body composition in response to GH therapy in AGHD(32). The major clinical predictors in our models were gender, body height, lean body mass, and serum insulin levels. In addition, genetic factors have been investigated as good candidates to explain the variability on GH responsiveness. We could not demonstrate any influence of polymorphisms of the GH receptor gene in the 12-month response to GH therapy in AGHD(33). In contrast, van der Klaauw and co-workers observed a higher increase in IGF-1 levels, higher increase in high-density lipoprotein cholesterol, lower decrease in total cholesterol and lower decrease in low-density lipoprotein cholesterol in patients bearing at least one exon 3 deleted-GH receptor (d3GHR) allele at short-term follow-up (1 yr), but not at long-term (5 yr), in their group of AGHD patients(34). Thus, there is a need for additional studies to define the importance of this polymorphism and others genetic factors in the clinical practice, using pharmacogenetics to optimize the therapeutic outcomes.
The benefits of GH therapy need to be weighed against the adverse events, which are dose-dependent and related to the metabolic effects of GH. In the initial studies of GH replacement, high doses based on body weight were employed and were associated with numerous side effects, specially related to fluid retention – such as paresthesias, joint stiffness, peripheral edema, arthralgia, myalgia and carpal tunnel syndrome(2). Subsequently, dosages were reduced, resulting in fewer adverse events and a safer profile. As a rule, GH therapy is, nowadays, a well-tolerated therapy for AGHD. However, special attention with older, heavier and female AGHD patients is still advisable, because they are more prone to develop adverse events(6-8). AGHD patients are insulin resistant –probably related to increased adiposity, reduced lean body mass, and impaired physical performance– which temporarily worsens when GH treatment is initiated(35). GH treatment appears to be associated with a slightly increased risk of impaired glucose tolerance and diabetes mellitus among AGHD. As a result, GH treatment needs to be prescribed with care and close monitoring in patients with a history of type 2 diabetes(6). GH treatment can also unmask a preexisting subclinical central hypothyroidism and secondary adrenal insufficiency and, therefore, free thyroxine and cortisol levels should be monitored during treatment with GH(36).
Despite the potential role of GH and IGF-1 in cell proliferation, there is no evidence of increased recurrence rates of intracranial or extracranial tumors among patients taking GH for AGHD(6-8). However, there are no published reports of long-term studies quantifying the increased risk, if any, of de novo malignancies, and GH should not be given to patients with active malignancy(6-8). On the other hand, monitoring for cancer development in GH-treated adults should be the same as for the general population. A slight excess risk of second neoplasia among children taking GH has been described, but no comparable data are available in adults(6, 37). Certainly, this represents an important area for future research.
WHICH THERAPEUTIC APPROACH SHOULD BE USED?
There is broad consensus that GH replacement therapy in adults should start with a low dose of GH, with subsequent dose titration to attain normal IGF-1 levels(6-8). At start, it is recommended a daily dose of 0.2 mg for young men and 0.3 mg for young women, while in older individuals a dose of 0.1 mg per day is more appropriate to initiate GH therapy (Figure 2). Patients should administer GH as a subcutaneous injection in the evening, at bedtime. Long-acting preparations might offer a possible alternative to daily subcutaneous injections in the future, but further studies are needed to better characterize the clinical role and pharmacokinetics of the long-acting preparations of GH(38, 39).

Figure 2. Therapeutic and follow-up procedures for GH replacement therapy in AGHD. BMI: body mass index; DXA: dual energy X-ray absorptiometry; QoL: quality of life; BMD: bone mineral density.
The dose of GH should be increased gradually, individualized, and guided by clinical and biochemic parameters. The main goal is to attain and maintain normal IGF-1 levels, preferably between the median and upper limit of the age-related normal range. However, in a study where a low and fixed dose of GH was given for one year, we have shown a positive and significant impact of GH therapy on body composition, even when IGF-1 levels were not normalized(40). The maintenance dose of GH shows considerable variation from person to person depending on various factors, such as age, sex, adiposity and hormone interactions, to name a few(30, 31). For example, women taking exogenous oral estrogen typically need higher doses of GH. Therefore, women who require exogenous estrogen should be preferably replaced with a non-oral route of administration. Moreover, any change in oral estrogen dose requires reevaluation of the GH dose(31, 36). The length of therapy is not well defined, but if benefits are being clearly achieved, there is no particular reason to stop treatment. On the other hand, if there are no apparent or objective benefits of treatment after at least 1 yr of follow-up, then the discontinuation of GH therapy seems appropriate(6).
There have been recommendations to use Qol evaluations as a tool for therapeutic approach in AGHD. Disease-specific questionnaire such as "Assessment of GHD in Adults (QoL-AGHDA)” and general questionnaires, such as "Psychological General Well-Being (PGWB)” and "Nottingham Health Profile (NHP)”, have been validated to measure a variety of health-related, economic, and social factors(41). However, in untreated AGHD, QoL evaluations have shown a high degree of variability, from normal to severe impairment, affecting mainly energy and vitality. As a rule, if the QoL is normal at baseline, no improvement should be expected with GH therapy, while the degree of improvement is generally proportional to the deviation from normality(6).
HOW TO FOLLOW THE PATIENTS DURING GH THERAPY?
Regular monitoring of the response to GH is essential (Figure 2). During GH dose adjustments, clinical assessments, side effects and serum IGF-1 levels should be monitored at intervals of 1 to 2 months. Thereafter, measurement of IGF-I is re-commended at least once a year to ascertain that levels are kept in the normal range and below the upper limit to avoid over-treatment.
Physicians should also measure the changes in body composition associated with GH treatment. Simple anthropometry, such as BMI and waist circumference, can be easily recorded, but ideally, dual energy X-ray absorptiometry (DXA) should be employed every year or two to better assess lean mass and body fat. DXA is also a good tool for detecting changes in bone mineral density, but these measurements have to be undertaken only two years after the initiation of GH therapy. Cardiovascular risk factors (particularly blood pressure, lipid profile and glucose) should be meas-ured every 6 or 12 months. Finally, clinicians are oriented to monitor health-related QoL outcomes –such as energy levels, partner satisfaction, sick days and vitality– by careful anamnesis, to ascert-ain a comprehensive view of the benefits of GH treatment(6-8). Disease-specific questionnaires are usually used as research tools and they need to be validated for country, ethnicity and language(41).
MORTALITY, LONGEVITY AND THE GH-IGF-1 AXIS
Several animal models with reduced GH and/or IGF-1 signaling have been shown to have extended life spans as compared to control siblings. Interestingly, the lack of GH and/or IGF-1 is able to keep the animals healthy and disease-free for longer periods and can alleviate specific age-related pathologies(42). In addition, a recent study in a large Brazilian pedigree with familial isolated GHD due to GHRH receptor mutation has suggested that longevity is not compromised by the absence of replacement GH therapy(43). In contrast, another study done in Switzerland suggested a reduced longevity in untreated patients with isolated GHD. However, the life span was not statistically different among controls and the 11 affected subjects who lived in two isolated Swiss valleys in the late 19th and early 20th centuries(44). Hence, most of the animal and human studies showing extended lifespan with good health in GH and IGF-1 deficiency create a paradox with the higher cardiovascular mortality associated with AGHD, which is characterized by increased abdominal adiposity, insulin resistance, cardiac dysfunctions and unfavorable lipid profile(2-8). A possible explanation for this apparent discrepancy is the relationship between the GH-IGF-1 axis and insulin sensitivity. The Brazilian patients with a specific genetic background showed GHD with very low, but measurable, GH levels, severe reduction of serum IGF-I and IGF-I binding protein type 3 (IGFBP-3), with a higher IGF-1/IGFBP-3 ratio, which might improve insulin action. Similarly, animal models with GH and/or IGF-1 deficiency and increased life span also demonstrate normal insulin sensitivity. In contrast, AGHD patients with other hypothalamic-pituitary diseases might have variable degrees of GH and IGF-1 secretion and insulin-resistance(42, 43). Thus, future studies should look more carefully on the relationship between the GH-IGF-1 axis, insulin sensitivity and glucose homeostasis.
CLOSING REMARKS
Several guidelines have been published in the last years which offer the clinicians a pragmatic approach to the management of AGHD(6-8). In addition, several reports in the last decade have added important information on the efficacy and safety of long-term follow-ups of AGHD patients treated or not treated with GH(45-51). Currently, adult patients with an evidence of any abnormality of the hypothalamic-pituitary area should be considered for evaluation of severe GHD and replacement therapy with GH. The ITT test is the most validated biochemical tool for diagnosis of AGHD, while the glucagon test is, nowadays, the best alternative for cases where ITT is unsuitable or contra-indicated. However, a low IGF-I level is sufficient for diagnosis, without the need for additional provocative testing, in patients with high probability of AGHD. GH therapy offers significant clinical benefits in body composition, exercise capacity, skeletal integrity, and QoL measurements, especially for patients with severe clinical and biochemical abnormalities. The adverse events and therapeutic risks are low. Protocols for GH dosing regimens and for monitoring the therapeutic outcomes are now well-established and easily available in the guidelines. Nevertheless, there is still a need for more research in several key areas, such as a better characterization of partial AGHD, factors influencing individual variation in the clinical response to GH therapy, long-term benefits and risks of GH therapy in relation to cardiovascular morbidity and mortality, insulin sensitivity, longevity, and tumor development, as well as the therapeutic potential of the long-acting preparations of GH. In the transition period, the educational process should start at the time of diagnosis in childhood, when the patient and the family are informed not only about the linear growth-stimulating effect of GH, but also on its life-long effects on body composition and metabol-ism(7, 8, 20). As recently discussed in a workshop involving paediatric and adult endocrinologists from several countries of Latin-America, the optimization of care during transition relies on the establishment of clinical programmes in specialized centers formed by multidisciplinary groups dedicated to the management of hypopituitarism and GHD patients(52).
1. Raben MS. Clinical use of human growth hormone. N Engl J Med 266: 82-86, 1962. [ Links ]
2. Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of adults with growth hormone deficiency: summary statement of the Growth Hormone Research Society workshop on adult. J Clin Endocrinol Metab 83: 379-381, 1998. [ Links ]
3. Rosén T, Bengtsson BA. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet 336(8710): 285-288, 1990. [ Links ]
4. Bulow B, Hagmar L, Eskilsson J, Erfurth EM. Hypopituitary females have high incidence of cardiovascular morbidity and an increased prevalence of cardiovascular risk factors. J Clin Endocrinol Metab 85: 574-584, 2000. [ Links ]
5. Tomlinson JW, Holden N, Hills R, Wheatley K, Clayton RN, Bates AS, Sheppard MC, Stewart PM. Premature mortality in 1014 patients with hypopituitarism. Lancet 357: 425-431, 2001. [ Links ]
6. Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Shalet SM, Vance ML; Endocrine Society's Clinical Guidelines Subcommittee, Stephens PA. Evaluation and treatment of adult growth hormone deficiency: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 91: 1621-1634, 2006. [ Links ]
7. Ho KK; 2007 GH Deficiency Consensus Workshop Participants. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol 157: 695-700, 2007. [ Links ]
8. Giustina A, Barkan A, Chanson P, Grossman A, Hoffman A, Ghigo E, Casanueva F, Colao A, Lamberts S, Sheppard M, Melmed S; Pituitary Society; European Neuroendocrine Association. Guidelines for the treatment of growth hormone excess and growth hormone deficiency in adults. J Endocrinol Inv 31: 820-838, 2008. [ Links ]
9. Corpas E, Harman SM, Blackman MR. Human growth hormone and human aging. Endocr Rev 14: 20-39, 1993. [ Links ]
10. Ho KY, Evans WS, Blizzard RM, Veldhuis JD, Merriam GR, Samojlik E, Furlanetto R, Rogol AD, Kaiser DL, Thorner MO. Effects of sex and age on the 24-hour profile of growth hormone secretion in man: importance of endogenous estradiol concentrations. J Clin Endocrinol Metab 64: 51-58, 1987. [ Links ]
11. Thorner MO. Statement by the Growth Hormone Research Society on the GH/IGF-I axis in extending health span. J Gerontol A Biol Sci Med Sci 64: 1039-1044, 2009. [ Links ]
12. Giordano R, Bonelli L, Marinazzo E, Ghigo E, Arvat E. Growth hormone treatment in human ageing: benefits and risks. Hormones (Athens) 7: 133-139, 2008. [ Links ]
13. Melmed S. Supplemental growth hormone in healthy adults: the endocrinologist's responsibility. Nat Clin Pract Endocrinol Metab 2: 119, 2006. [ Links ]
14. Vance ML. Can growth hormone prevent aging? N Engl J Med 348: 779-780, 2003. [ Links ]
15. Tauber M, Moulin P, Pienkowski C, Jouret B, Rochiccioli P. Growth hormone (GH) retesting and auxological data in 131 GH-deficient patients after completion of treatment. J Clin Endocrinol Metab 82: 352-356, 1997. [ Links ]
16. Lacerda CS, Carvalho JR, Boguszewski MCS, Boguszewski CL. Reavaliação Diagnóstica da deficiência de hormônio de crescimento (DGH) na fase de transição entre adolescência e vida adulta. Arq Bras Endocrinol Metab 52(Suppl 1): S60, 2008. [ Links ]
17. Shalet SM. Partial growth hormone deficiency in adults; should we be looking for it? Clin Endocrinol (Oxf). 2010 Apr 23. [ Epub ahead of print] [ Links ].
18. Yuen KC, Biller BM, Molitch ME, Cook DM. Is lack of recombinant growth hormone (GH)-releasing hormone in the United States a setback or time to consider glucagon testing for adult GH deficiency? J Clin Endocrinol Metab 94: 2702-2707, 2009. [ Links ]
19. Conceição FL, da Costa e Silva A, Leal Costa AJ, Vaisman M. Glucagon stimulation test for the diagnosis of GH deficiency in adults. J Endocrinol Invest 26: 1065-1070, 2003. [ Links ]
20. Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M; European Society of Paediatric Endocrinology. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol 152: 165-170, 2005. [ Links ]
21. Maghnie M, Aimaretti G, Bellone S, Bona G, Bellone J, Baldelli R, de Sanctis C, Gargantini L, Gastaldi R, Ghizzoni L, Secco A, Tinelli C, Ghigo E. Diagnosis of GH deficiency in the transition period: accuracy of insulin tolerance test and insulin-like growth factor-I measurement. Eur J Endocrinol 152: 589-596, 2005. [ Links ]
22. Bidlingmaier M. Pitfalls of insulin-like growth factor I assays. Horm Res 71(Suppl 1): 30-33, 2009. [ Links ]
23. Clayton P, Gleeson H, Monson J, Popovic V, Shalet SM, Christiansen JS. Growth hormone replacement throughout life: insights into age-related responses to treatment. Growth Horm IGF Res 17: 369-382, 2007. [ Links ]
24. Maison P, Griffin S, Nicoue-Beglah M, Haddad N, Balkau B, Chanson P. Impact of growth hormone (GH) treatment on cardiovascular risk factors in GH-deficient adults: a Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. J Clin Endocrinol Metab 89: 2192-2199, 2004. [ Links ]
25. Widdowson WM, Gibney J. The effect of growth hormone replacement on exercise capacity in patients with GH deficiency: a metaanalysis. J Clin Endocrinol Metab 93: 4413-4417, 2008. [ Links ]
26. Cook DM. Shouldn't adults with growth hormone deficiency be offered growth hormone replacement therapy? Ann Intern Med 137: 197-201, 2002. [ Links ]
27. Isley WL. Growth hormone therapy for adults: not ready for prime time? Ann Intern Med 137: 190-196, 2002. [ Links ]
28. Frohman L. Controversy about treatment of growth hormone-deficient adults: a commentary. Ann Intern Med 137: 202-204, 2002. [ Links ]
29. Danilowicz K, Bruno OD, Manavela M, Gomez RM, Barkan A. Correction of cortisol overreplacement ameliorates morbidities in patients with hypopituitarism: a pilot study. Pituitary 11: 279-285, 2008. [ Links ]
30. Johannsson G, Bjarnason R, Bramnert M, Carlsson LM, Degerblad M, Manhem P, Rosen T, Thorén M, Bengtsson BA. The individual responsiveness to growth hormone (GH) treatment in GH-deficient adults is dependent on the level of GH-binding protein, body mass index, age, and gender. J Clin Endocrinol Metab 81: 1575-1581, 1996. [ Links ]
31. Ho KK, Gibney J, Johannsson G, Wolthers T. Regulating of growth hormone sensitivity by sex steroids: implications for therapy. Front Horm Res 35: 115-128, 2006. [ Links ]
32. Barbosa EJ, Koranyi J, Filipsson H, Bengtsson BA, Boguszewski CL, Johannsson G. Models to predict changes in serum IGF1 and body composition in response to GH replacement therapy in GH-deficient adults. Eur J Endocrinol 162: 869-878, 2010. [ Links ]
33. Barbosa EJ, Palming J, Glad CA, Filipsson H, Koranyi J, Bengtsson BA, Carlsson LM, Boguszewski CL, Johannsson G. Influence of the exon 3-deleted/full-length growth hormone (GH) receptor polymorphism on the response to GH replacement therapy in adults with severe GH deficiency. J Clin Endocrinol Metab 94: 639-644, 2009. [ Links ]
34. van der Klaauw AA, van der Straaten T, Baak-Pablo R, Biermasz NR, Guchelaar HJ, Pereira AM, Smit JW, Romijn JA. Influence of the d3-growth hormone (GH) receptor isoform on short-term and long-term treatment response to GH replacement in GH-deficient adults. J Clin Endocrinol Metab 93: 2828-2834, 2008. [ Links ]
35. Møller N, Jørgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 30: 152-177, 2009. [ Links ]
36. Filipsson H, Johannsson G. GH replacement in adults: interactions with other pituitary hormone deficiencies and replacement therapies. Eur J Endocrinol 161(Suppl 1): S85-95, 2009. [ Links ]
37. Ergun-Longmire B, Mertens AC, Mitby P, Qin J, Heller G, Shi W, Yasui Y, Robison LL, Sklar CA. Growth hormone treatment and risk of second neoplasms in the childhood cancer survivor. J Clin Endocrinol Metab 91: 3494-3498, 2006. [ Links ]
38. Clemmons DR. Long-acting forms of growth hormone-releasing hormone and growth hormone: effects in normal volunteers and adults with growth hormone deficiency. Horm Res 68(Suppl 5): 178-181, 2007. [ Links ]
39. Rasmussen MH, Bysted BV, Anderson TW, Klitgaard T, Madsen J. Pegylated Long-Acting Human Growth Hormone Is Well-Tolerated in Healthy Subjects and Possesses a Potential Once-Weekly Pharmacokinetic and Pharmacodynamic Treatment Profile. J Clin Endocrinol Metab. 2010 Apr 28. [ Epub ahead of print] [ Links ].
40. Boguszewski CL, Meister LH, Zaninelli DC, Radominski RB. One year of GH replacement therapy with a fixed low-dose regimen improves body composition, bone mineral density and lipid profile of GH-deficient adults. Eur J Endocrinol 152: 67-75, 2005. [ Links ]
41. Koltowska-Häggström M, Mattsson AF, Shalet SM. Assessment of quality of life in adult patients with GH deficiency: KIMS contribution to clinical practice and pharmacoeconomic evaluations. Eur J Endocrinol 161(Suppl 1): S51-S64, 2009. [ Links ]
42. Berryman DE, Christiansen JS, Johannsson G, Thorner MO, Kopchick JJ. Role of the GH/IGF-1 axis in lifespan and healthspan: lessons from animal models. Growth Horm IGF Res 18: 455-471, 2008. [ Links ]
43. Aguiar-Oliveira MH, Oliveira FT, Pereira RM, Oliveira CR, Blackford A, Valenca EH, Santos EG, Gois-Junior MB, Meneguz-Moreno RA, Araujo VP, Oliveira-Neto LA, Almeida RP, Santos MA, Farias NT, Silveira DC, Cabral GW, Calazans FR, Seabra JD, Lopes TF, Rodrigues EO, Porto LA, Oliveira IP, Melo EV, Martari M, Salvatori R. Longevity in untreated congenital growth hormone deficiency due to a homozygous mutation in the GHRH receptor gene. J Clin Endocrinol Metab 95: 714-721, 2010. [ Links ]
44. Besson A, Salemi S, Gallati S, Jenal A, Horn R, Mullis PS, Mullis PE. Reduced longevity in untreated patients with isolated growth hormone deficiency. J Clin Endocrinol Metab 88: 3664-3667, 2003. [ Links ]
45. Zaninelli DC, Meister LH, Radominski RB, Borba VZ, Souza AM, Boguszewski CL. Efficacy, safety and compliance of long-term growth hormone (GH) replacement therapy in adults with GH deficiency. Arq Bras Endocrinol Metabol 52: 879-888, 2008. [ Links ]
46. Cenci MC, Soares DV, Spina LD, de Lima Oliveira Brasil RR, Lobo PM, Mansur VA, Gold J, Michmacher E, Vaisman M, Conceição FL. Effects of 5 years of growth hormone (GH) replacement therapy on cardiac parameters and physical performance in adults with GH deficiency. Pituitary 12: 322-329, 2009. [ Links ]
47. Götherström G, Bengtsson BA, Bosaeus I, Johannsson G, Svensson J. Ten-year GH replacement increases bone mineral density in hypopituitary patients with adult onset GH deficiency. Eur J Endocrinol 156: 55-64, 2007. [ Links ]
48. Götherström G, Elbornsson M, Stibrant-Sunnerhagen K, Bengtsson BA, Johannsson G, Svensson J. Ten years of growth hormone (GH) replacement normalizes muscle strength in GH-deficient adults. J Clin Endocrinol Metab 94: 809-816, 2009. [ Links ]
49. Olsson DS, Buchfelder M, Schlaffer S, Bengtsson BA, Jakobsson KE, Johannsson G, Nilsson AG. Comparing progression of non-functioning pituitary adenomas in hypopituitarism patients with and without long-term GH replacement therapy. Eur J Endocrinol 161: 663-669, 2009. [ Links ]
50. Arwert LI, Roos JC, Lips P, Twisk JW, Manoliu RA, Drent ML. Effects of 10 years of growth hormone (GH) replacement therapy in adult GH-deficient men. Clin Endocrinol (Oxf) 63: 310-316, 2005. [ Links ]
51. Gilchrist FJ, Murray RD, Shalet SM. The effect of long-term untreated growth hormone deficiency (GHD) and 9 years of GH replacement on the quality of life (QoL) of GH-deficient adults. Clin Endocrinol (Oxf) 57: 363-370, 2002. [ Links ]
52. Lanes R, Boguszewski CL, Calzada R, Cassorla F, Fideleff H, Boquete H. Growth hormone deficiency: transition from adolescence to adulthood. Highlights from a Latin-American Serono Symposia International Foundation Conference. J Pediatr Endocrinol Metab 23: 225-233, 2010. [ Links ]

