










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista argentina de endocrinología y metabolismo
versión On-line ISSN 1851-3034
Rev. argent. endocrinol. metab. vol.47 no.4 Ciudad Autónoma de Buenos Aires oct./dic. 2010
TRABAJO DE CASUÍSTICA
Síndrome de Klinefelter en las distintas edades: experiencia multicéntrica
Klinefelter Syndrome at differents ages: multicentric experience
Pacenza N.1, Pasqualini T.2, Gottlieb S.3, Knoblovits P.4, Costanzo P.4, Stewart Usher J.5, Rey R.3, Martínez M.6, Aszpis S.6
1Servicio de Endocrinología y Metabolismo, Unidad Asistencial "Dr. César Milstein", Buenos Aires.
2Sección Endocrinología, Crecimiento y Desarrollo, Departamento de Pediatría, Hospital Italiano, Buenos Aires.
3Centro de Investigaciones Endocrinológicas (CEDIE, CONICET), División de Endocrinología, Hospital de Niños Dr. Ricardo Gutiérrez, Buenos Aires.
4Servicio de Endocrinología, Metabolismo y Medicina Nuclear, Hospital Italiano, Buenos Aires.
5Consultorio de Endocrinología, Centro Médico Haedo, Haedo (Provincia de Buenos Aires).
6Sección Andrología, División Endocrinología, Hospital Durand, Buenos Aires
Correspondencia: Dr. Néstor Pacenza. Dirección postal: Av. Díaz Vélez 3889, (1200) CABA, Argentina. e-mail: napacenza@yahoo.com.ar
Resumen
El Síndrome de Klinefelter (SK) es la anormalidad cromosómica más frecuente en los varones, con una prevalencia estimada de 1:600 recién nacidos.
El objetivo de este trabajo fue establecer las distintas características de presentación del SK a distintas edades, incluyendo signos y síntomas clínicos, parámetros de laboratorio y otros exámenes complementarios.
La franja etaria más frecuente de diagnóstico de SK fue entre los 11 y 20 años (46,8%). En 4 casos el diagnóstico fue prenatal. Los motivos de consulta más frecuentes en forma global fueron la presencia de testículos pequeños, infertilidad y criptorquidia. El cariotipo más prevalente fue el clásico 47,XXY (83,7%), seguido del mosaico 47,XXY/46,XY (7,1%).
El promedio de talla de nuestros pacientes prepuberales no mostró diferencia con la población general. Por otro lado, los pacientes puberales presentaron un promedio de talla significativamente más alto, hallándose alrededor de 1 SDS. Hubo correlación entre la edad y el SDS de talla. La media de talla de los adultos fue 178,8 ± 9,0 cm; se observó un 62,5% de sobrepeso/obesidad (IMC ≥ 25,0 kg/m2).
El 50% de nuestros pacientes con SK menores de 18 años presentaron trastornos neurocognitivos. El hallazgo clínico más frecuente entre los pacientes prepuberales fue la criptorquidia. En los puberales las consultas y hallazgos clínicos más frecuentes fueron: testículos pequeños, criptorquidia y ginecomastia. Todos nuestros pacientes en estadio de Tanner igual o mayor de III presentaron testículos más pequeños para su grado de desarrollo. Los valores de FSH y LH fueron normales en los pacientes prepuberales y comenzaron a aumentar en la pubertad.
Los adultos consultaron más frecuentemente por hipotrofia testicular, infertilidad y en menor grado ginecomastia. Todos los pacientes presentaron testículos hipotróficos, con una mediana de volumen testicular de 3,5 (1-8) ml. El 56,4% presentaron función sexual normal; el resto tuvo algún tipo de disfunción sexual. La testosterona total (TT) fue normal en 45% de los pacientes, con descenso consistente con la edad, donde todos los pacientes mayores de 40 años presentaron TT subnormal. El 10,7% de los pacientes que efectuaron espermograma tuvo oligospermia severa, el resto presentó azoospermia. La densitometría ósea fue anormal en el 46,4% de los adultos estudiados. Sin embargo, no hubo diferencias significativas en la prevalencia de osteopenia y osteoporosis entre los pacientes con TT normal o subnormal.
Los autores declaran no poseer conflictos de interés.
Palabras clave: Klinefelter; Hipogonadismo; 47,XXY; Testosterona
Abstract
Klinefelter syndrome (KS) is the most common chromosomal aberration among men, with an estimated prevalence of 1:600 newborns. It is an X chromosome polysomy, with X disomy being the most common variant (47,XXY).
The aim of this study was to establish the characteristics of KS presentation at different ages, including signs and symptoms, laboratory parameters and other diagnostic tests.
The diagnosis of KS was more frequent in the age group between 11 and 20 years (46.8%). Most of the patients (83.7%) showed the classic 47,XXY karyotype and 7.1% showed a 47,XXY/46,XY mosaicism.
While mean prepubertal height was not different from the control population, it was significantly higher at puberty.
Patients consulted most frequently for small testes, infertility and cryptorchidism. In four cases the diagnosis was prenatal. 50% of our patients younger than 18 years presented neurocognitive disorders. The more frequent clinical findings were cryptorchidism in prepubertal patients; small testes, cryptorchidism and gynecomastia in pubertal patients. All our patients in Tanner stage III or more presented small testes. FSH and LH levels were normal in prepubertal patients and increased abnormally at puberty.
On the other hand, most adults consulted for small testes, infertility and gynecomastia. 43.6% of patients had decreased libido, sexual and/or ejaculatory dysfunction.
In adults average height (178.8 ± 9.0 cm) and weight (83.6 ± 21.0 kg), were higher than in the normal population, however 8 patients (19%) had a height less tan 170 cm. There was 62.5% of overweight / obesity (BMI ≥ 25.0 kg/m2) in the whole group of adult patients. 35.2% had eunuchoid proportions. All patients had testicular hypotrophc, with a median testicular volume of 3.5 ml (range 1-8 ml). Total testosterone (TT) levels were normal in 45% of adult patients, showing significant correlation with age. All patients aged 40 or more years had subnormal TT levels. In patients who underwent semen analysis, severe oligospermia and azoospermia were found in 10.7% and 89.3% respectively. Bone mineral densitometry showed low bone mass in 46.4% of cases. No significant differences in the prevalence of osteopenia and osteoporosis were observed among patients with normal or subnormal TT.
No financial conflicts of interest exist.
Key words: Klinefelter; Hypogonadism; 47,XXY; Testosterone
INTRODUCCIÓN
Klinefelter, Reifenstein y Albright describieron en 1942 un síndrome hallado en 9 varones con ginecomastia, testículos pequeños, azoospermia y aumento de FSH(1). Más tarde, Jacobs y Strong establecen el cariotipo característico de este síndrome al describir un paciente con síndrome de Klinefelter (SK) con cariotipo 47,XXY(2).
La constitución cromosómica es, en la mayoría de los casos, la disomía del X (47,XXY) en forma homogénea (SK no mosaico) y, en una minoría, en forma no homogénea, es decir que se presenta en mosaico con otras líneas celulares normales o anormales (SK mosaico). También se han observado casos, aunque de escasa incidencia, de 48,XXXY, 49,XXXXY e incluso 48,XXYY ya sean puros o en mosaicos(3).
Se han descripto fenotipos variables, dependiendo del grado de deficiencia androgénica del paciente. En algunos, la deficiente virilización comienza a temprana edad por severa disfunción del eje gonadal, con niveles subnormales de testosterona, gonadotrofinas elevadas y signos de insuficiencia androgénica. En el otro extremo están los que pasan desapercibidos hasta la vida adulta y consultan generalmente por infertilidad.
Por su parte, el diagnóstico en niños es menos frecuente hasta la pubertad, siendo los signos de alarma la criptorquidia y los trastornos neurocognitivos(4).
El objetivo general de este trabajo es establecer las características de las diferentes formas de presentación del SK a distintas edades, incluyendo signos y síntomas clínicos, así como parámetros de laboratorio y otros exámenes complementarios.
PACIENTES Y MÉTODOS
Estudio observacional retrospectivo multicéntrico, en el que se incluyeron los datos obtenidos de las historias clínicas de pacientes con diagnóstico confirmado de SK (polisomía del cromosoma X y al menos un cromosoma Y, ya sea en línea pura o en mosaico), con conformidad de los Comités de Ética y Docencia e Investigación de cada centro participante.
El análisis se realizó en forma global y por subgrupos, de acuerdo a la edad, en varones prepúberes (menores de 10 años), en etapa puberal (10 a 17,9 años) y adultos (a partir de los 18 años).
Se consideraron las siguientes variables: edad al diagnóstico, presencia o antecedente de criptorquidia, ginecomastia, trastornos neurocognitivos (trastornos de aprendizaje y/o de conducta) y otros datos que surgieran de la anamnesis, examen físico, niveles hormonales [hormona folículoestimulante (FSH), hormona luteinizante (LH), testosterona total (TT), prolactina (PRL), estradiol (E2), perfil tiroideo, inhibina B, hormona antimülleriana (AMH)], colesterol total (CT), triglicéridos (TG), HDL y LDL colesterol, glucemia, espermograma y densitometría ósea (DMO).
Expresión de resultados: En cada subgrupo etario, los resultados se presentan como: a) Variables cualitativas: frecuencia de presentación al momento de la consulta; b) Variables cuantitativas: media ± DE o mediana y rango según corresponda.
Estadística: Las medias ± DE fueron comparadas mediante el test de Student para datos no apareados y las proporciones fueron comparadas por la prueba de chi2, utilizando el software GraphPad InStat, versión 3.01.
La prevalencia de los distintos hallazgos se informa sobre el número total de pacientes en los cuales el dato estuvo registrado.
RESULTADOS
Se analizaron las historias clínicas de 98 pacientes con diagnóstico confirmado de SK, 54 mayores y 44 menores de 18 años de edad (18 pacientes menores de 10 años y 26 de 10 a 17,9 años).
En cuanto a la edad de diagnóstico de SK, la mediana de edad fue 17,0 años (IC 95%: 15,9 - 20,4 años), con un rango de 0-54 años. La franja etaria más prevalente fue entre los 11 y los 20 años (44/94 pacientes, 46,8%) (Figura 1). El diagnóstico de SK se efectuó en etapa prenatal en 4 casos.

Figura 1. Edad al diagnóstico de SK (n=94). En 4 pacientes no pudo ser establecida la edad al diagnóstico.
Los motivos de consulta más frecuentes fueron la presencia de testículos pequeños, esterilidad y criptorquidia (Tabla I).
Los cariotipos más frecuentes fueron: el clásico 47,XXY (83,7%) y el mosaico 47,XXY/46,XY (7,1%) (Tabla II). Los 4 casos de diagnóstico prenatal tuvieron cariotipo clásico 47,XXY.
TABLA I. Motivos de consulta de los pacientes con SK (n=102)*
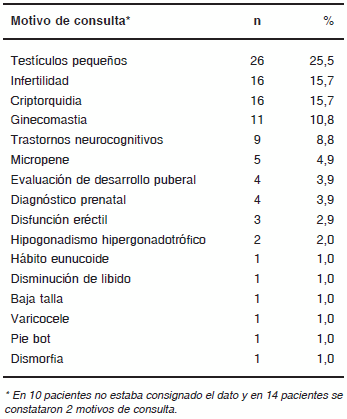
TABLA II. Cariotipos de los pacientes intervinientes en el estudio (n=98)

Anamnesis y Examen Físico
Menores de 18 años
La mediana de edad al momento del diagnóstico de SK de los 44 pacientes menores de 18 años fue de 13,2 (0 - 17,5) años, 2,2 (0 - 9,5) años en los prepúberes, incluyendo los 4 de diagnóstico prenatal y 14,4 (10,1 - 17,5) años en los púberes.
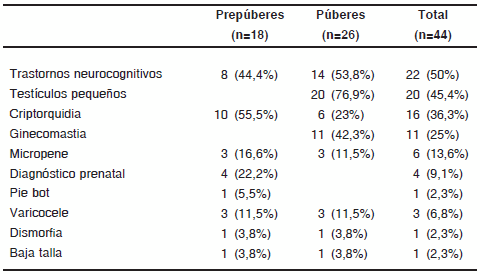
Si bien 9 pacientes (20,4%) consultaron por trastornos neurocognitivos, por interrogatorio se pudo determinar que el 50% de los menores de 18 años presentaban estos trastornos, que incluyeron: retardo madurativo y en la adquisición del lenguaje, dificultades escolares y trastornos de conducta (Tabla III).
TABLA III. Anamnesis y hallazgos clínicos en pacientes menores de 18 años con SK
En 29 pacientes se pudo determinar la edad parental (mediana, rango) al momento de la concepción: 29 (17 - 44) años la edad materna y 33 (20-55) años la paterna.
La evaluación antropométrica se pudo completar en 15 pacientes prepuberales y en 25 puberales (Tabla IV). El índice de masa corporal (IMC) no fue significativamente diferente entre los pacientes prepuberales y puberales. La diferencia de las medias de talla entre los pacientes prepuberales y puberales fue significativa, presentando estos últimos una media mayor de 1 SDS (p<0.0001). Por otro lado, se halló una correlación significativa entre el SDS de talla y la edad (n=40, r=0.304, p<0.05). La talla por encima de 2 SDS fue observada en uno de los pacientes prepuberales y en cinco de los puberales.
TABLA IV. Datos antropométricos de pacientes prepuberales*

La presencia de criptorquidia se constató en 16 de los 44 pacientes (36,3%), que resultó ser el hallazgo clínico más frecuente en los pacientes prepuberales (Tabla III). En cuanto a su tratamiento, 9 (56,2%) requirieron cirugía.
En los pacientes prepuberales, el volumen testicular escrotal varió entre 1 y 2 ml, aunque 3 pacientes presentaron un volumen menor de 1 ml en forma bilateral.
El volumen testicular disminuido fue el hallazgo clínico más frecuente en los pacientes puberales (76,9%). La mediana del volumen testicular fue 3,7 (1-8) ml. Todos los pacientes con grado de desarrollo igual o mayor a estadío III de Tanner presentaron un volumen testicular más pequeño para su grado de desarrollo.
La ginecomastia fue el segundo hallazgo más frecuente en los pacientes puberales, evidenciándose en el 42,3% de los casos.
Se constató micropene y varicocele en 6 y 3 pacientes respectivamente (Tabla III).
Mayores de 18 años
En la población evaluada a una edad igual o mayor de 18 años, la mediana de edad al diagnóstico de SK fue de 23 (5-54) años.
Los motivos de consulta más frecuentes al diagnóstico en este grupo fueron: testículos de pequeño tamaño e infertilidad, representando casi el 70% entre los 2 (Tabla V).
TABLA V. Motivo de consulta en pacientes adultos con SK (n=46)*

De los registros clínicos examinados no se pudieron obtener los datos suficientes para informar la edad de los padres al momento de la concepción de los pacientes, edad de comienzo de la pubertad, trastornos cognitivos ni orientación sexual.
Seis de 46 pacientes (13%) tuvieron antecedente de criptorquidia.
En 26 pacientes se obtuvieron datos sobre el nivel educativo: 25 completaron el ciclo primario y 14 el secundario; 11 pacientes continuaron estudios terciarios. Las alteraciones de la función sexual se especifican en la Tabla VI.
TABLA VI. Función sexual en adultos con SK

La media de talla fue de 178,8 ± 9,0 cm. Ocho pacientes presentaron talla menor de 170 cm (19%). El peso promedio fue 83,6 ± 21,0 kg (Tabla VII). La mediana de IMC fue de 25,9 (rango 17,2 - 51,1) kg/m2, hallándose 19 pacientes (47,5%) con sobrepeso (IMC 25,0 - 29,9 kg/m2) y 6 pacientes (15%) con obesidad (IMC ≥ 30,0 kg/m2) (Tabla VII).
TABLA VII. Datos antropométricos de pacientes adultos con SK

Diecinueve pacientes (35,2%) presentaron hábito eunucoide. Ninguno de los pacientes examinados presentó micropene al examen.
Todos los pacientes presentaron testículos hipotróficos. La mediana del volumen testicular fue 3,5 (1-8) ml.
La prevalencia de varicocele fue 23,3% (7/30); 4 del lado izquierdo, 1 bilateral y en 2 no fue consignada la lateralidad.
Se evidenció ginecomastia en 15 pacientes (31,2%). Doce (80%) fueron bilaterales.
En 5 pacientes se observaron defectos anatómicos: pectum excavatum (n=2), hallux más largo (n=1), pie cavo varo (n=1), quinto metacarpiano corto (n=1), escoliosis (n=1) (1 paciente tuvo 2 defectos anatómicos).
Se encontraron enfermedades endocrino-metabólicas concomitantes en 5 pacientes: hipotiroidismo (n=1), hipertiroidismo (n=1), prolactinoma (n=1), diabetes mellitus tipo 2 (n=1) y obesidad mórbida (n=1).
Laboratorio
Menores de 18 años
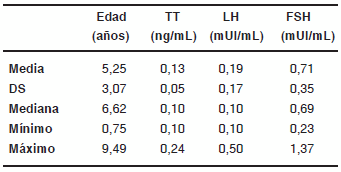
En 13 de los pacientes menores de 10 años que pudieron ser estudiados, los valores de FSH, LH y TT fueron normales (Tabla VIII). También resultaron normales los valores de AMH e Inhibina B.
TABLA VIII. Valores de TT, LH y FSH en pacientes menores de 10 años con SK (n=13)
En etapa puberal fueron estudiados 28 pacientes; tres de ellos habían sido diagnosticados en etapa prepuberal. Se hallaron niveles elevados de FSH en 20 y de LH en 16 pacientes. Se halló una correlación negativa entre los niveles de gonadotrofinas y la edad cronológica (FSH y edad: r= -0.62, p<0.001; LH y edad: r= -0.59, p<0.001), observándose valores elevados de FSH a partir de los 12,8 años y de LH a partir de los 13,8 años (estadio III de Tanner) (Figuras 2 y 3). No se halló correlación entre edad cronológica y valores de TT (Figura 4). Los valores de AMH e inhibina B disminuyeron por debajo de lo normal a partir del estadio III de Tanner.
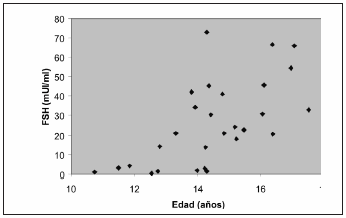
Figura 2. Relación de FSH con edad en púberes con SK (n=28) r= 0.62, p<0.001

Figura 3. Relación de LH con edad en púberes con SK (n=28) r= 0.59, p<0.001

Figura 4. Relación de TT con edad en púberes con SK (n=28) r= 0.36, p=NS
Siete pacientes comenzaron tratamiento con testosterona antes de los 18 años de edad.
Mayores de 18 años
El 45,1% (23/51) de los pacientes mantenía un nivel sérico de TT dentro del rango normal al momento de la evaluación, según los valores de referencia del método de laboratorio utilizado en cada caso. Hubo una diferencia significativa entre las edades de los pacientes con TT subnormal y la de los pacientes con TT normal: 32,8 ± 10,5 versus 24,7 ± 6,4 años, p<0.002. Además, se halló una correlación negativa entre la edad y los niveles de TT (r= -0.5, p<0.001) (Figura 5). Tuvieron TT normal el 69% de los pacientes entre 18 y 20 años, el 40% entre 20,1 y 30 años, el 28,5% entre 30,1 y 40 años, no hallando ningún caso de TT normal en los mayores de 40 años.

Figura 5. Relación de TT con edad en adultos con SK (n=51) r= -0.5, p<0.001
Treinta y nueve de 47 pacientes (83%) tuvieron niveles de LH por encima del rango normal. Cuatro de los 8 pacientes con LH normal presentaban TT por debajo del rango normal. El 100% de los pacientes tuvo FSH elevada (12,1 - 74,9 mUI/mL). Once pacientes presentaron valores de PRL por encima del rango normal (33%); en uno de ellos se halló un adenoma prolactínico. El 6,2% (2/32) tenía E2 elevado. La TSH fue normal en los 24 pacientes en que fue evaluada.
Diez de 25 pacientes (40%) evidenciaron un CT mayor de 200 mg/dL (8 de ellos con TT subnormal). Tres de 20 (15%) reflejaron TG > 170 mg/dL, 10 de 20 (50%) un LDL > 130 mg/dL y 9 de 19 (47,4%) pacientes un HDL < 45 mg/dL. Por otro lado, ninguno de los 22 pacientes evaluados tuvo niveles de glucemia en ayunas > 126 mg/dL.
Espermograma
De los 28 pacientes que realizaron espermograma, 25 presentaron azoospermia y 3 tuvieron oligospermia severa: uno (47,XXY) con 900.000 espermatozoides/ml, otro (47,XXY/46,XY) con 200.000 espermatozoides/ml y el tercero (47,XXY) con 1 espermatozoide cada 40 campos.
Densitometría ósea
Las características de las DMO de región lumbar (L2-L4) y cadera (cuello de fémur) efectuadas en los pacientes con SK se expresan en la Tabla IX.
TABLA IX : Densitometría ósea (DMO) en pacientes adultos con SK

Trece de los 28 pacientes que efectuaron DMO de columna lumbar y/o de cadera (46,4%), presentaron baja masa ósea (T score < -1.0). Hubo 2 pacientes (7,4%) con osteoporosis (T score < -2.5) de columna y no hubo pacientes con osteoporosis de cadera. No pudieron analizarse los Z score dado los escasos registros del dato en las historias clínicas.
Ocho de los 13 pacientes con baja masa ósea (61,5%) tenían valores de TT subnormal, mientras que, de los 15 pacientes con DMO normal, 7 presentaron TT por debajo del rango normal (46,7%) (p=ns). Por otro lado, si analizamos el total de pacientes que tenían TT subnormal, tampoco hubo diferencias significativas entre los que presentaban o no baja masa ósea. Es decir, no hubo correlación entre los niveles de TT y la DMO en estos pacientes.
DISCUSIÓN
El Síndrome de Klinefelter (SK) es una aneuploidía de los cromosomas sexuales con polisomía del X, con una prevalencia estimada de 1 en 600 recién nacidos varones, siendo la anormalidad cromosómica más frecuente(5).
Los cariotipos más frecuentes en nuestra población fueron: el clásico 47,XXY (83,7%) y el mosaico 47,XXY/46,XY (7,1%), muy similar a lo previamente descripto, en donde el cariotipo clásico se halló en el 80-90% de los pacientes con SK(5,6).
Si bien el fenotipo clásico del SK es ampliamente conocido, muchos pacientes afectados sólo presentan síntomas discretos, por lo que este desorden suele ser subdiagnosticado. Alrededor del 25% de los pacientes con SK se diagnostican en edad adulta y el 10% es diagnosticado antes de la pubertad(5,7). En nuestra casuística, el 46,8% de los pacientes con SK fue diagnosticado entre los 11 y los 20 años (Figura 1).
La mediana de edad de los padres al momento de la concepción fue 29 (17- 44) años para la edad materna y 33 (20 - 55) años para la edad paterna. Se ha descripto que el SK es más prevalente a medida que aumenta la edad materna y posiblemente también la edad paterna(5,8).
El 50% de nuestros pacientes con SK menores de 18 años presentaron trastornos neurocognitivos que incluyeron: retardo madurativo, retardo en la adquisición del lenguaje, dificultades escolares y trastornos de conducta. Estudios prospectivos han demostrado que los varones con SK presentan dificultades en la adquisición del lenguaje además de otras dificultades académicas y psicológicas que pueden reducirse con el diagnóstico temprano, la guía hacia los padres y maestros y el adecuado manejo médico(4,9,10). Por lo tanto, la presencia de trastornos neurocognitivos, en especial el retraso en la adquisición del lenguaje, en un varón prepuberal, con o sin criptorquidia, debe alertar al pediatra para eventual solicitud de un cariotipo.
A pesar que se describe aumento de talla en los pacientes con SK entre los 5 y 8 años de edad, el promedio de talla de nuestros pacientes prepuberales no mostró diferencia con la población general(11,12). Este hallazgo contrasta con lo publicado recientemente, en donde se halló que los niños prepuberales ya presentaban talla elevada(13). Por otro lado, los pacientes puberales presentaron un promedio de talla significativamente más alto que el de los prepuberales, hallándose alrededor de 1 SDS. Hubo una correlación entre la edad y el SDS de talla. Se cree que la talla alta del SK está relacionada principalmente a la presencia de una copia extra del gen SHOX en los cromosomas X e Y, y en menor medida a un retardo de la fusión epifisaria secundaria a niveles reducidos de testosterona y estradiol(14). El hallazgo de obesidad o sobrepeso no fue universal en nuestros pacientes menores de 18 años.
El promedio de talla y peso de los pacientes adultos con SK (178,8 cm y 83,5 kg, respectivamente) fue mayor a la observada en la población argentina de 18 años de edad, en quienes el percentilo 50 de talla adulta fue 172,8 ± 6,8 cm y el percentilo 50 de peso fue 65,7 ± 10,4 kg para la hemidistribución superior(15). Sin embargo, 8 pacientes adultos (19%) presentaron una talla menor de 170 (161-169) cm. Es claro que frente a las características fenotípicas clásicas debe pensarse en el SK aún en paciente con talla normal.
El hallazgo clínico más frecuente entre los pacientes prepuberales fue la criptorquidia (55,5%). El 23% de los pacientes puberales también la presentaron. Nuestro hallazgo es similar al observado en un estudio de 29 pacientes pediátricos (16 prepuberales y 13 puberales) donde se halló criptorquidia en el 69% de los prepúberes (5 bilaterales y 6 unilaterales) y en el 31% de los puberales (1 bilateral y 3 unilateral)(16). Cabe aclarar que la mayor prevalencia de criptorquidia en prepuberales puede estar relacionada con el estudio del cariotipo a raíz de la propia criptorquidia. Por otro lado, en un estudio que evaluó las alteraciones genéticas asociadas con criptorquidia, se halló un 1,3% de alteraciones cromosómicas en el grupo de pacientes con criptorquidia versus 0,3% en el grupo control. Cuando se estudiaron los pacientes con criptorquidia bilateral, la frecuencia de alteración cromosómica fue de 4,2%. Todas las alteraciones cromosómicas halladas en el grupo criptórquido fue del tipo SK, ya sea puro o en mosaico(17). Dado que la criptorquidia es más frecuente en el SK que en los normales, debería solicitarse estudios genéticos en todos los pacientes con criptorquidia(17,18).
Nosotros hallamos un tamaño testicular disminuido en nuestros pacientes, aún desde la edad prepuberal. Algunos estudios previos también describieron tamaño testicular disminuido en edad prepuberal(19), mientras que otros lo hallaron recién en etapa puberal(12). El motivo de consulta y hallazgo clínico más frecuente en nuestros pacientes puberales fue la presencia de menor volumen testicular que se halló en el 76,9% de los pacientes. De hecho, todos nuestros pacientes en estadio de Tanner igual o mayor de III presentaron testículos más pequeños para su grado de desarrollo. Parecería que la reducción del tamaño testicular en etapa prepuberal podría ser secundaria a una reducción del tamaño de los túbulos seminíferos con reducción o ausencia de espermatogonias. Una minoría de los túbulos suelen ser normales, siendo la mayoría más pequeños, con células de Sertoli indiferenciadas(20). La naturaleza focal del proceso degenerativo ya es evidente, dado que los escasos túbulos seminíferos que contienen espermatogonias están rodeados por túbulos seminíferos que contienen sólo células de Sertoli(21,22). La diferenciación de células germinales no suele estar retrasada, ya que los gonocitos maduran a espermatogonias sin retraso, pero el estudio histológico testicular muestra que existe arresto en estadío de espermatocito primario sin presencia de espermatocitos paquiténicos. Parece que en el SK, la espermatogonia tiene dificultad para entrar en meiosis y al momento del inicio de la pubertad entra en apoptosis(21-23). Las células de Leydig y Sertoli podrían estar preservadas hasta el inicio de la pubertad, donde las células de Sertoli comienzan a degenerar y las células de Leydig se vuelven hiperplásicas(22).
El síndrome originalmente descripto por Klinefelter(1) no se reconoce en el recién nacido ya que estos pacientes suelen tener diferenciación sexual masculina completa. Por lo tanto, éste no es un diagnóstico diferencial a tener en cuenta en el recién nacido con genitales ambiguos, a pesar que se ha descripto presencia de micropene, hallazgo presente en 6 de nuestros pacientes. Sin embargo, Lee y col. describieron anormalidades genitales en 7 pacientes con SK, sugiriendo que es importante conocer esta asociación y reconocer al SK como una de las causas de genitales anormales al nacer(24).
Al igual que nuestros hallazgos, se ha descripto niveles normales de FSH y LH hasta el comienzo de la pubertad(19,22-27). Desde la mitad de la pubertad en adelante, los pacientes con SK muestran un aumento gradual de FSH y LH. Los niveles de FSH aumentan primero y en forma mayor que los de LH(19,22,25,27). Nosotros pudimos observar que, igual a lo previamente descripto, los niveles de FSH aumentan después de los 12 años, llegando a niveles más elevados que los de LH. Esta última hormona aumenta un poco más tardíamente, alrededor de los 14 años.
En nuestro grupo de pacientes prepuberales, se halló una TT dentro del rango normal. Aksglaede y col. hallaron un valor de TT y testosterona libre significativamente mayor en infantes con SK que en controles normales(28). Durante la pubertad, los niveles de TT, después de un aumento inicial, muestran una meseta y permanecen en general dentro de los valores normales bajos(19,25-27,30). Nuestros hallazgos fueron similares, observando un aumento peripuberal normal de TT, con niveles que luego quedaban en un valor promedio de 2,35 ng/mL. Sólo 5 de 28 pacientes puberales superaron los 3 ng/mL. Sin embargo, estos niveles de TT fueron suficientes para permitir el inicio y progreso de la pubertad normal con el desarrollo adecuado de los caracteres sexuales secundarios(23,25).
Siete pacientes comenzaron tratamiento con testosterona antes de los 18 años de edad. El desarrollo de una relativa deficiencia de testosterona desde la mitad de la pubertad se corresponde con una disfunción de las células de Leydig que se traduce en una disminución de los niveles no sólo de TT, sino también del factor insulinosímil 3 (INSL3) y en un aumento de LH(23,25,26). Esto coincide con los hallazgos histomorfométricos y análisis inmunohistoquímicos que observan hiperplasia de células de Leydig y fibrosis del intersticio que se desarrollan con la edad y la inmunoexpresión del receptor de andrógenos que indica disminución de la acción androgénica(21,22). A pesar que parecería necesaria la suplementación androgénica en el SK desde la mitad de la pubertad, no existen todavía estudios placebo-control que muestren el beneficio de este tratamiento temprano en cuanto al efecto positivo sobre parámetros de conducta y cognitivos. Por otro lado, aparentemente la capacidad de obtener espermatozoides es menor en aquellos que recibieron previamente terapia androgénica sustitutiva(31). Entonces, el potencial mantenimiento de la fertilidad debe ser balanceado contra los beneficios potenciales de la testosterona.
Los motivos de consulta de los pacientes con SK en edad adulta, coinciden con lo descripto por otros autores. Kamischke y col., en 85 pacientes con SK, hallaron como motivos de consulta preponderantes: esterilidad (37), hipogonadismo (11) y en menor grado ginecomastia (2)(32).
Hemos encontrado testículos hipotróficos en todos los pacientes adultos, con un volumen medio de 3,5 ml. Lenz y col., en un grupo de 18 pacientes con SK, describen un volumen medio de 5 ml para ambos testículos(33). Otros, hallan una media de 4,7 ml bilateral de volumen testicular, en un grupo de 85 pacientes, y consideran que el volumen testicular total es el parámetro que más ayuda a distinguir los pacientes con SK de otros hipogonadismos hipergonadotróficos con azoospermia(32).
La prevalencia de varicocele en nuestros pacientes adultos fue 23,3%, similar a la prevalencia descripta en la población general(34). El 13% de los pacientes (6/46) tuvo antecedente de criptorquidia. Kamischke y col. hallaron un 17% (14/84) de pacientes con SK con dicho antecedente(32).
El 45,1% (23/51) de los pacientes tenía un nivel sérico de TT dentro del rango normal, según los valores de referencia de cada laboratorio. La media de edad de los pacientes con TT subnormal (n=28) fue 32,8 ± 10,5 años, mientras que la media de edad de los pacientes con TT normal fue 24,7 ± 6,4 años (p<0.002). Otro grupo encontró valores de TT normal (> 3,0 ng/mL) en 33/40 pacientes con SK (82,5%)(31). Los valores de TT parecen disminuir con la edad más tempranamente que en la población general, probablemente por su menor reserva testicular(35).
Se hallaron niveles elevados de CT en el 40%, de TG en el 15% y de LDL en el 50% y subnormales de HDL en el 47,7% de los pacientes evaluados. Bojesen y col. hallaron niveles significativamente más altos de CT, LDL y TG y más bajos de HDL en pacientes con SK no tratados comparados con los normales, valores que no se modificaron sustancialmente con el tratamiento androgénico(36).
Veinticinco de los 28 pacientes que efectuaron espermograma presentaron azoospermia (89,3%). Tres tuvieron oligospermia severa (10,7%), dos 47,XXY y un mosaico 47,XXY/46,XY. Kamischke y col. hallaron espermatozoides en 4 de 57 pacientes (7%); los cuatro fueron: 47,XXY(32).
En cuanto a la DMO de nuestros pacientes adultos, el 46,4% presentó baja masa ósea (T score < -1), con sólo 2 casos de osteoporosis de columna y ninguno de cadera. Seo y col. describen una media de DMO significativamente menor en pacientes con SK comparado con controles normales(37). Además estos autores hallaron una falta de correlación lineal entre los niveles de TT y la DMO, similar a nuestros hallazgos. La mayoría de los estudios que evalúan DMO en pacientes con SK demuestran una menor masa ósea respecto al grupo control, aunque es raro encontrar casos de osteoporosis marcada(38,39).
Agradecimientos
A todos los colegas que pusieron a disposición las historias clínicas de sus pacientes para este análisis.
1. Klinefelter HF, Reifenstein EC, Albright F. Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism, and increased excretion of follicle-stimulating hormone. J Clin Endocrinol Metab 2:615-627, 1942. [ Links ]
2. Jacobs PA, Strong J.A. A case of human intersexuality having a possible XXY sex-determining mechanism. Nature 183:302-303, 1959. [ Links ]
3. Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics 96:672-682, 1995. [ Links ]
4. Ratcliffe S. Long term outcome in children of sex chromosome abnormalities. Arch Dis Child 80:192-195, 1999. [ Links ]
5. Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab 88:622-626, 2003. [ Links ]
6. Lanfranco F, Kamischke A, Zitzmann M, Nieschlag E. Klinefelter's Syndrome. Lancet 364:273-283, 2004. [ Links ]
7. Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat Diagn 17:363-368, 1997. [ Links ]
8. Lowe X, Eskenazi B, Nelson DO, Kidd S, Alme A, Wyrobek AJ. Frequency of XY sperm increases with age in fathers of boys with Klinefelter syndrome. Am J Hum Genet 69:1046-1054, 2001. [ Links ]
9. Visootsak J, Aylstock M, Graham JM Jr. Klinefelter syndrome and its variants: an update and review for the primary pediatrician. Clin Pediatr (Phila) 40:639-651, 2001. [ Links ]
10. Aksglaede L, Skakkebaek NE, Juul A. Abnormal sex chromosome constitution and longitudinal growth: serum levels of insulin-like growth factor (IGF)-I, IGF binding protein-3, luteinizing hormone, and testosterone in 109 males with 47,XXY, 47,XYY, or sex-determining region of the Y chromosome (SRY)-positive 46,XX karyotypes. J Clin Endocrinol Metab 93:169-176, 2008. [ Links ]
11. Lejarraga H, Orfila G. Estándares de peso y estatura para niños y niñas argentinos después del nacimiento hasta la madurez. Arch Arg Pediatr 85:209-222, 1987. [ Links ]
12. Stewart DA, Bailey JD, Netley CT, Park E. Growth, development, and behavioral outcome from mid-adolescence to adulthood in subjects with chromosome aneuploidy: the Toronto Study. Birth Defects Orig Artic Ser 26:131-188, 1990. [ Links ]
13. Zeger MP, Zinn AR, Lahlou N, Ramos P, Kowal K, Samanago-Sprouse C, Ross JL. Effect of ascertainment and genetic features on the phenotype of Klinefelter syndrome. J Pediatr 152:716-722, 2008. [ Links ]
14. Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch J, Winkelmann M, Nordsiek G, Heinrich U, Breuning MH, Ranke MB, Rosenthal A, Ogata T, Rappold GA. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet 16:54-63, 1997. [ Links ]
15. Lejarraga H, Abeyá Gilardon EO, Andrade JH, Boffi Boggero H. Evaluación del peso y la talla en 88.861 varones de 18 años de la República Argentina (1987). Arch Arg Pediatr 89:185-192, 1991. [ Links ]
16. Bastida MG, Rey RA, Bergadá I, Baldecarrás P, Andreone L, del Rey G, Boywitt A, Ropelato MG, Cassinelli H, Arcari A, Campo S, Gottlieb S. Establishment of testicular endocrine function impairment during childhood and puberty in boys with Klinefelter síndrome. Clin Endocrinol (Oxf) 67:863-870, 2007. [ Links ]
17. Ferlin A, Zuccarello D, Zuccarello B, Chirico MR, Zanon GF, Foresta C. Genetic alterations associated with cryptorchidism. JAMA 300:2271-2276, 2008. [ Links ]
18. Bergadá C, Farías NE, Romero de Behar BM, Cullen M. Abnormal sex chromatin pattern in cryptorchidism, girls with short stature and other endocrine patients. Helv Paediatr Acta 24:372-377, 1969. [ Links ]
19. Salbenblatt JA, Bender BG, Puck MH, Robinson A, Faiman C, Winter JS. Pituitary-gonadal function in Klinefelter syndrome before and during puberty. Pediatr Res 19:82-86, 1985. [ Links ]
20. Muller J, Skakkebaek NE, Ratcliffe SG. Quantified testicular histology in boys with sex chromosome abnormalities. Int J Androl 18:57-62, 1995. [ Links ]
21. Wikström AM, Hoei-Hansen CE, Dunkel L, Raj-pert-De Meyts E. Immunoexpression of androgen receptor and nine markers of maturation in the testes of adolescent boys with Klinefelter syndrome: evidence for degeneration of germ cells at the onset of meiosis. J Clin Endocrinol Metab 92:714-719, 2007. [ Links ]
22. Wikström AM, Raivio T, Hadziselimovic F, Wikström S, Tuuri T, Dunkel L. Klinefelter syndrome in adolescence: onset of puberty is associated with accelerated germ cell depletion. J Clin Endocrinol Metab 89:2263-2270, 2004. [ Links ]
23. Wikström AM, Dunkel L. Testicular Function in Klinefelter Syndrome. Horm Res 69:317-326, 2008. [ Links ]
24. Lee YS, Cheng AW, Ahmed SF, Shaw NJ, Hughes IA. Genital anomalies in Klinefelter's syndrome. Horm Res 68:150-155, 2007. [ Links ]
25. Wikström AM, Dunkel L, Wickman S, Norjavaara E, Ankarberg-Lindgren C, Raivio T. Are adolescent boys with Klinefelter syndrome androgen deficient? A longitudinal study of Finnish 47,XXY boys. Pediatr Res 59:854-859, 2006. [ Links ]
26. Wikström AM, Bay K, Hero M, Andersson AM, Dunkel L. Serum INSL3 levels during puberty in healthy boys and boys with Klinefelter syndrome. J Clin Endocrinol Metab 91:4705-4708, 2006. [ Links ]
27. Topper E, Dickerman Z, Prager-Lewin R, Kaufman H, Maimon Z, Laron Z. Puberty in 24 patients with Klinefelter syndrome. Eur J Pediatr 139:8-12, 1982. [ Links ]
28. Aksglaede L, Petersen J, Main K, Skakkebæk N, Juul A. High normal testosterone levels in infants with non-mosaic Klinefelter's syndrome. Eur J Endocrinol 157:345-350, 2007. [ Links ]
29. Christiansen P, Andersson AM, Skakkebaek NE. Longitudinal studies of inhibin B levels in boys and young adults with Klinefelter syndrome. J Clin Endocrinol Metab 88:888-891, 2003. [ Links ]
30. Winter JS. Androgen therapy in Klinefelter syndrome during adolescence. Birth Defects Orig Artic Ser 26:235-245, 1990. [ Links ]
31. Schiff JD, Palermo GD, Veeck LL, Goldstein M, Rosenwaks Z, Schlegel PN. Success of testicular sperm injection and intracytoplasmic sperm injection in men with Klinefelter syndrome. J Clin Endocrinol Metab 90:6263-6267, 2005. [ Links ]
32. Kamischke A, Baumgardt A, Horst J, Nieschlag E. Clinical and diagnostic features of patients with suspected Klinefelter syndrome. J Androl 24:41-48, 2003. [ Links ]
33. Lenz P, Luetjens CM, Kamischke A, Kühnert B, Kennerknecht I, Nieschlag E. Mosaic status in lymphcytes of infertile men with or without Klinefelter´s syndrome. Human Reprod 20:1248-1255, 2005. [ Links ]
34. Saypol DC. Varicocele. J Androl 2:61-71, 1981. [ Links ]
35. Paduch DA, Bolyakov A, Cohen P, Travis A. Reproduction in men with Klinefelter syndrome: the past, the present and the future. Semin Reprod Med 27:137-148, 2009. [ Links ]
36. Bojesen A, Kristensen K, Birkebaek NH, Fedder J, Mosekilde L, Bennett P, Laurberg P, Frystyk J, Flyvbjerg A, Christiansen JS, Gravholt CH. The metabolic syndrome is frequent in Klinefelter's syndrome and is associated with abdominal obesity and hypogonadism. Diabetes Care 29:1591-1598, 2006. [ Links ]
37. Seo JT, Lee JS, Oh TH, Joo KJ. The clinical significance of bone mineral density and testosterone levels in Korean man with non-mosaic Klinefelter´s syndrome. BJU Int 99:141-146, 2007. [ Links ]
38. Bojesen A, Gravholt CH. Klinefelter syndrome in clinical practice. Nat Clin Pract Urol 4:192-204, 2007. [ Links ]
39. Aszpis S, Gottlieb S, Knoblovits P, Pacenza N, Pasqualini T, Rey R, Stewart Usher J. Síndrome de Klinefelter: Viejos y nuevos conceptos. Rev Argent Endocrinol Metab 43: 22-39, 2006. [ Links ]

