










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkActa toxicológica argentina
versión On-line ISSN 1851-3743
Acta toxicol. argent. vol.26 no.1 Ciudad Autónoma de Buenos Aires mayo 2018
REVISIÓN
Hidroarsenicismo: mecanismos de acción asociados a la toxicidad del arsénico
Hydroarsenicism: Mechanisms of action related to arsenic toxicity
Olmos, Valentina*; Ridolfi, Adriana S.
Universidad de Buenos Aires, Facultad de Farmacia y Bioquímica. Catedra de Toxicología y Química Legal. Junín 956, 7º, Ciudad Autónoma de Buenos Aires.
Recibido: 15 de mayo de 2018
Aceptado: 30 de mayo de 2018
Resumen
La exposición crónica al arsénico (As) inorgánico a través del agua de bebida da lugar al desarrollo de la enfermedad conocida como hidroarsenicismo. Esta enfermedad presenta sintomatología característica, sin embargo, para la mayoría de los efectos tóxicos que produce del As aún no se conoce en detalle el mecanismo de acción tóxica. Los mecanismos moleculares de acción del arsenito (unión a grupos sulfhidrilos) y del arseniato (sustitución del fosfato) están bien identificados, sin embargo, las consecuencias a nivel subcelular, celular, tisular y orgánico de esos mecanismos todavía presentan muchos huecos por llenar. A nivel subcelular y celular, la generación de especies reactivas de oxígeno (ERO) y de nitrógeno (ERN) son los mecanismos de acción tóxica del As más estudiados últimamente. Se los ha vinculado con la diferenciación y proliferación de queratinocitos, con la disfunción endotelial, con la resistencia a la insulina, con la inducción de peroxidación lipídica en hígado, de necrosis tubular renal y con cambios en la expresión del receptor estrogénico. Por último, la respuesta celular a proteínas no plegadas (como consecuencia del estrés del retículo endoplásmico) podría ser un mecanismo para explicar la afectación de la inmunidad humoral y la celular.
Palabras clave: Arsénico; Mecanismos de acción, Hidroarsenicismo; Estrés oxidativo.
Abstract
Chronic exposure to inorganic arsenic (As) through drinking water leads to the development of the disease known as hydroarsenicism. This disease presents characteristic symptomatology but the mechanisms underlying most of the toxic effects produced by As are not fully understand. The molecular mechanisms of action of arsenite (binding to sulfhydryl groups) and arsenate (phosphate substitution) are well identified, however, the consequences at the subcellular, cellular, tissue and organic levels of these mechanisms still have many gaps to fill. At the subcellular and cellular level, the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) are the most studied mechanisms of toxic action. They have been linked to the differentiation and proliferation of keratinocytes, endothelial dysfunction, insulin resistance, induction of lipid peroxidation in the liver, renal tubular necrosis and changes in the expression of estrogen receptor. Finally, the cellular response to unfolded proteins (as a consequence of the stress of the endoplasmic reticulum) could be a mechanism to explain the affectation of humoral and cellular immunity.
Keywords: Arsenic; Mechanisms of action, Hydroarsenicism; Oxidative stress.
Introducción
La exposición crónica durante años al As a través del agua de bebida da lugar al desarrollo de la enfermedad conocida como hidroarsenicismo. Esta enfermedad presenta sintomatología característica, sin embargo, para la mayoría de los efectos tóxicos que produce del As aún no se conoce en detalle el mecanismo de acción tóxica.
Las propiedades químicas del As fueron las que abrieron el camino hacia los potenciales mecanismos moleculares de acción tóxica. Las primeras propuestas de mecanismos de acción aparecieron a principios del siglo XX y estuvieron relacionadas a las propiedades químicas conocidas, hasta ese momento, de los compuestos de As. Por ejemplo: la afinidad por los grupos sulfhidrilos del arsenito (AsIII) y la similitud estructural del arseniato (AsV) con el fosfato. Un factor importante a la hora de analizar los mecanismos de acción y la toxicidad de los compuestos del As además de su valencia (trivalente o pentavalente), se relaciona con el tipo de compuesto (por ejemplo: metilado o no metilado).
Actualmente se investigan varios mecanismos de acción como responsables de los efectos adversos y tóxicos del As y, además, se cree que estos mecanismos podrían ser interdependientes. Las líneas de investigación van desde las acciones a nivel molecular de los compuestos de As, siguiendo con sus efectos a nivel subcelular, celular, tisular, orgánico y sistémico.
Para una mejor comprensión de la toxicodinamia del As, esta revisión se organizó en tres niveles:
1. Mecanismos de acción moleculares y efectos subcelulares y celulares
2. Efectos en tejidos, órganos y sistemas y los mecanismos de acción involucrados
3. Mecanismos de acción asociados a carcinogénesis
1. Mecanismos de acción moleculares y efectos subcelulares y celulares
1.1. Unión a grupos sulfhidrilos
La primera evidencia de acción tóxica a través de este mecanismo se demostró sobre el complejo enzimático piruvato deshidrogenasa (PDHc). El PDHc está compuesto por tres enzimas: piruvato deshidrogenasa (PDH), dihidrolipoil transacetilasa (DLT) y dihidrolipoil deshidrogenasa (DLD), y por varios cofactores enzimáticos (Figura 1). Este complejo cataliza la producción de nicotinamida adenina dinucleótido reducido (NADH), acetil coenzima A (ACoA) y flavina adenina dinucleótido reducido (FADH2). Se comprobó que el arsenito y los arsenicales metilados trivalentes se unen al ácido lipoico, una molécula con grupos sulfhidrilos vecinales, que participa del PDHc como cofactor enzimático (Figura 1).

Figura 1. Reacciones catalizadas por el complejo piruvato deshidrogenasa (PDHc) e inhibición provocada por el arsenito. CO2: dióxido de carbono, Acetil-TPP: acetil tiamina pirofosfato, TPP: tiamina pirofosfato, PDH: piruvato deshidrogenasa, DLT: dihidrolipoil transacetilasa, CoASH: coenzima A, Acetil CoA: acetil coenzima A, DLD: dihidrolipoil deshidrogenasa, FAD+: flavina adenina dinucleótido oxidado, FADH2: flavina adenina dinucleótido reducido, NAD+: nicotinamida adenina dinucleótido oxidado, NADH: nicotinamida adenina dinucleótido reducido.
De esta manera se interrumpe la producción de NADH, ACoA y FADH2. La ACoA es un precursor de los intermediarios del ciclo del ácido cítrico y el NADH y el FADH2 se utilizan en la síntesis de adenosina trifosfato (ATP) por la vía de la fosforilación oxidativa. La inhibición del PDHc interrumpe el ciclo del ácido cítrico y la producción de ATP llevando al daño y a la muerte celular.
Además, a través del mismo mecanismo de unión a grupos sulfhidrilos, el arsenito y los intermediarios metabólicos trivalentes del As inhiben casi 200 proteínas y enzimas. Entre ellas, se pueden mencionar, la glutatión reductasa, la tiorredoxina, la tiorredoxina reductasa, las ADN ligasas, las enzimas reparadoras de ADN y la piruvato kinasa (Hughes 2002, Shen y col. 2013). La inhibición se produce por unión del As a grupos sulfhidrilos de cisteínas localizadas en posiciones clave de la estructura proteica, de los sitios activos enzimáticos o por unión a cofactores enzimáticos ditiólicos. En muchos de los casos, los intermediaros metabólicos metilados trivalentes del As, como el ácido monometilarsenioso (MMAIII) y el ácido dimetil arsenioso (DMAIII), son inhibidores más potentes que el arsenito, y todos ellos, a su vez, son inhibidores mucho más potentes que los intermediarios metabólicos pentavalentes ácido monometilarsónico (MMAV) y el ácido dimetil arsínico (DMAV).
1.2. Sustitución del grupo fosfato
El ácido arsénico (AsO4H3) y el ácido fosfórico (PO4H3) tienen una estructura química similar y sus constantes de disociación también son similares, razón por la cual el arseniato puede sustituir al fosfato en muchas reacciones bioquímicas. En estas reacciones, el arseniato se une a grupos hidroxilos dando como resultado ésteres de arseniato. Sin embargo, la unión éster formada por el arseniato tiene mayor longitud que la formada por el fosfato, lo cual lleva a que los ésteres de arseniato sean más lábiles (menos estables) y se hidrolicen más fácilmente (Hughes y col. 2011).
El arseniato desacopla la formación de ATP in vitro por un mecanismo que se conoce como arsenolisis. La arsenolisis puede ocurrir en el proceso de la glucólisis y en la fosforilación oxidativa. En la glucólisis, el arseniato forma el 1-arseno, 3-fosfoglicerato (Figura 2) y en la fosforilación oxidativa el arseniato puede acoplarse a la adenosina difosfato (ADP) para formar el ADP-arseniato (Figura 3).
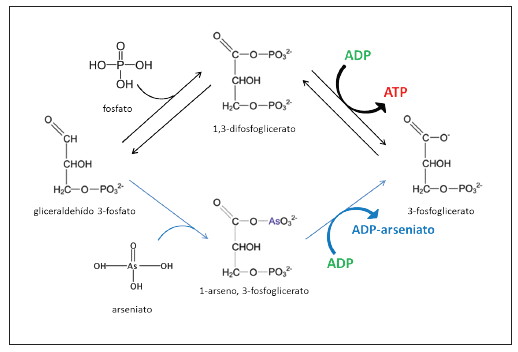
Figura 2. Esquema de la incorporación de fosfato (arriba) o arseniato (abajo) en el pasaje de gliceraldehído 3-fosfato a 3-fosfoglicerato de la glucólisis. ADP: adenosina difosfato, ATP: adenosina trifosfato
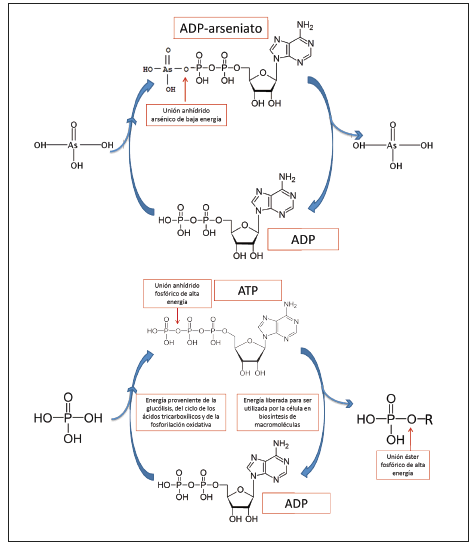
Figura 3. Formación de adenosina trifosfato (ATP) a partir de adenosina difosfato (ADP) (arriba) y formación de ADP-arseniato a partir de ADP (abajo). ADP: adenosina difosfato, ATP: adenosina trifosfato
1.3. Generación de especies reactivas de oxígeno y de nitrógeno
La generación de especies reactivas de oxígeno (ERO) y de nitrógeno (ERN) son los mecanismos de acción tóxica del As más estudiados últimamente (Flora 2011, Hughes y col. 2011). En estudios in vitro e in vivo se identificaron varias ERO: anión superóxido, radical hidroxilo, peróxido de hidrógeno, entre otras, como resultantes de la acción del As. El mecanismo de producción de ERO por parte del As no está totalmente establecido, podría tratarse de un mecanismo directo de inducción de peroxidación lipídica por parte del arsenito o sus intermediarios metabólicos, podrían generarse como consecuencia de la inhibición de la actividad de enzimas como la glutatión reductasa y la tiorredoxina reductasa, las cuales son parte de la defensa que tiene la célula contra el daño oxidativo o podrían generarse debido a la estimulación de las enzimas NADH o NADPH oxidasas (Hughes 2002, Samikkanu y col. 2003, Hei y Filipic 2004, Flora 2011, Hughes y col. 2011, Jomova y col. 2011, Dodmane y col.2013).
Otro mecanismo que se postula para la formación de ERO es a través de la liberación del hierro (Fe) de la ferritina. El ácido dimetil arsenioso (DMAIII) aumenta significativamente la liberación de Fe de la ferritina. El ácido dimetil arsínico (DMAV) produce el mismo efecto solo en presencia de un reductor como el ácido ascórbico. El arseniato, el arsenito, el monometil arsenioso (MMAIII) y el monometil arsónico (MMAV) no parecen actuar por este mecanismo (Hughes 2002).
2. Efectos en tejidos, órganos y sistemas. Mecanismos de acción involucrados
2.1 Efectos sobre la piel
Las lesiones cutáneas son las manifestaciones más conocidas de la exposición crónica al As. El tipo de lesiones que predominan son queratosis, hiperpigmentación y neoplasias. Sin embargo, los mecanismos por los cuales estas lesiones se originan, continúan siendo investigados. Desde hace más de 30 años se vienen proponiendo varios mecanismos celulares, subcelulares y moleculares que podrían estar vinculados al desarrollo de las alteraciones dérmicas que provoca la exposición crónica al As. Entre ellos se pueden mencionar:
• La supresión de la expresión de proteínas clave en el proceso de diferenciación de los queratinocitos epidérmicos (involucrina, transglutaminasas, proteína rica en prolina 1 y filagrina, entre otras) (Kachinskas y col. 1994, Jessen y col 2001). Todas estas proteínas y enzimas participan en la formación de la epidermis, particularmente en la diferenciación del queratinocito para formar el estrato córneo de la piel.
• La inducción de la sobreexpresión de factores de crecimiento (factor transformante de crecimiento alfa, factor estimulante de colonias de granulocitos y macrófagos y receptor del factor de crecimiento epidérmico, entre otros) puede ocurrir en queratinocitos, como respuesta a estímulos am bientales (Germolec y col. 1996).
• Inducción de la proliferación de queratinocitos mediada por la regulación de factores de transcripción como el factor nuclear kappa beta (NF-κβ) y el factor activador de proteínas-1 (AP-1) (Liao y col. 2004). Pi y col. (2008) mostraron que la activación del factor de transcripción nuclear Nrf2 en queratocitos humanos, en respuesta a estresores oxidativos, incrementa la secreción de múltiples biomarcadores de transformación maligna, entre ellos la metaloproteinasa- 9 (MMP-9) y citoqueratinas, que contribuyen a la carcinogénesis de piel por exposición a arsénico. La citotoxicidad del As inorgánico y sus metabolitos metilados trivalentes en queratocitos se relaciona con respuestas al estrés mediadas por la activación de Nrf2, interferón y p53 (Dodmane y col. 2013).
• Inducción de apoptosis (mediada por receptor) en queratinocitos: con aumento en la expresión del receptor Fas/FasL y con la subsecuente activación de las caspasas 8 (iniciadora) y 3 (ejecutora) (Liao y col. 2004).
• La activación de la respuesta mediada por proteínas no plegadas (UPR, por sus siglas en inglés, unfolded protein response). Esta activación se produciría como consecuencia de la generación de estrés oxidativo afectando particularmente al retículo endoplásmico, y daría como resultado una respuesta inflamatoria por parte de la piel (Li y col. 2011). La respuesta a proteínas no plegadas (UPR) es la respuesta del retículo endoplasmático al estrés oxidativo. Se desencadenan una serie de eventos que comienzan con la acumulación de proteínas no plegadas dentro del RE, su liberación al citoplasma celular y la activación de vías proteolíticas que intentan recuperar la homeostasis celular. Si ésta no se recupera, la UPR lleva a la apoptosis.
Estos mecanismos pueden ocurrir simultáneamente aunque se ha descripto que algunos podrían desencadenarse a concentraciones biológicas de As mayores que otros.
2.2. Efectos sobre el sistema cardiovascular
El aumento en la generación de ERO y de ERN, debidos a la exposición crónica al As, está propuesto como uno de los mecanismos relacionados con la aparición de efectos cardiovasculares (Singh y col. 2011). Las células del endotelio vascular y las adyacentes del músculo liso vascular son blanco del arsenito y sus intermediarios metabólicos. Algunos autores atribuyen el aumento en la generación de ERO y de ERN, a la estimulación de la NADPH oxidasa, presente en la membrana plasmática de estas células (Edwards y col. 2013, Ellinsworth 2015). Este desbalance redox provoca lo que se conoce como disfunción endotelial (desbalance de los factores de contracción y relajación del endotelio), la cual es el marcador de efecto temprano de patologías como la ateroesclerosis, la hipertensión o la enfermedad vascular periférica.
2.3. Efectos sobre el metabolismo de la glucosa
La arsenolisis y la formación del ADP-arseniato enlentecen el metabolismo de la glucosa, bloquean la obtención de energía e interfieren con la secreción (ATP-dependiente) de insulina (Singh y col. 2011). También, mediante la unión a grupos sulfhidrilos, el As puede bloquear la acción de la insulina, del receptor de insulina, del transportador de glucosa y de las enzimas del metabolismo de la glucosa (Singh y col. 2011). A su vez, la formación de ERO puede llevar a la sobreexpresión de estresores involucrados en el desarrollo de resistencia a la insulina (Singh y col. 2011). En conjunto, estas acciones pueden favorecer el desarrollo de diabetes mellitus en personas expuestas en forma crónica al As.
2.4. Neurotoxicidad
Se ha descripto que la exposición crónica al As a través del agua de bebida produce alteraciones en el desarrollo neurológico y cognitivo de los niños (Rodríguez Barranco y col. 2013), las cuales incluyen la afectación del coeficiente intelectual a escala completa, alteraciones en la memoria y la atención. También se ha asociado la exposición crónica al As con el desarrollo de la enfermedad de Parkinson y de polineuropatías símil síndrome de Guillan- Barré (Vahidnia y col. 2007, Singh y col. 2011).
Los mecanismos de acción asociados a estos efectos no están dilucidados. Sin embargo, varios trabajos proponen hipótesis sobre mecanismos de acción. El mecanismo más mencionado es a través de la generación de estrés oxidativo por aumento en la producción de ERO y disminución en la capacidad antioxidante de la célula neuronal (Singh y col. 2011, Prakash y col. 2015). Lu y colaboradores (2014) describieron algunos mecanismos de inducción de apoptosis neuronal como el mediado por la kinasa c-Jun N-terminal y la kinasa relacionada a la señalización extracelular (c-Jun N-terminal y extracellular signalrelated kinases, JNK y ERK por sus siglas en inglés) o el mediado por las proteínas vinculadas al estrés del retículo endoplásmico como la proteína reguladora de glucosa 78 y la proteína homóloga a C/ERP (glucose-regulated protein, C/EBP homologue protein, GRP 78 y CHOP, por sus siglas en inglés).
Otros mecanismos incluyen la alteración del metabolismo de neurotransmisores (dopamina, noradrenalina y 5-hidroxitriptamina), la modulación del calcio intracelular y la sobreexpresión y la activación de calpaínas (proteasas tiólicas citosólicas dependientes de calcio) que pueden actuar sobre las proteínas del citoesqueleto axonal, alterándolo (Florea y col. 2005, Vahidnia y col. 2007, 2008, Rocha y col. 2011, Prakash y col. 2016). La elevada acumulación de especies de arsénico en la pituitaria, respecto a otras áreas cerebrales podría explicar los efectos neuroendocrinos asociados con la exposición a As (Sánchez- Peña y col. 2010). Es probable que estos potenciales mecanismos de acción del As sean interdependientes y que, además, se vinculen con los mecanismos por los cuales el As altera el metabolismo basal de todas las células (glucólisis y fosforilación oxidativa).
2.5. Hepatotoxicidad
El As induce daño hepático caracterizado por hepatomegalia, fibrosis portal (no-cirrótica) y, en algunos casos, alteración de las enzimas aspartato aminotransferasa y alanino aminotransferasa (Guha Mazumder 2005, Singh y col. 2011). Otras alteraciones hepáticas, como la hipertensión portal (no cirrótica), se describieron en casos de pacientes que recibieron tratamiento crónico con trióxido de arsénico.
Los mecanismos vinculados a este tipo de daño se relacionan con un aumento en la generación de ERO, ya sea por aumento de la peroxidación lipídica, por disminución de los niveles de glutatión y tiorredoxina o a través de la inhibición de enzimas involucradas en la defensa antioxidante de la célula (catalasa, glutatión peroxidasa, tiorredoxina reductasa y glutatión reductasa, entre otras) (Santra y col. 2000, Singh y col. 2011, Li y col. 2018). Está descripto que el estrés oxidativo puede llevar a la apoptosis de células hepáticas (posiblemente a través de la regulación por incremento de proteínas pro-apoptóticas) (Singh y col. 2011, Li y col. 2018). La apoptosis podría verse favorecida en los casos de deficiencia de folato o de hiperhomocisteinemia.
2.6. Nefrotoxicidad
El As se acumula en el riñón, y así ejerce su efecto tóxico, especialmente en el tubo contorneado proximal. El daño renal se caracteriza por necrosis tubular con aumento de la urea y la creatinina en sangre. El mecanismo propuesto es a través de la generación de estrés oxidativo (Singh y col. 2011), debido a la inhibición de la actividad de la glutatión peroxidasa y la glutatión S-transferasa (Matos y col. 2013), e induciendo la sobreexpresión del gen codificante de la hemooxigenasa 1 (Sasaki y col. 2007).
2.7. Toxicidad sobre el tracto gastrointestinal
Los efectos tóxicos y los mecanismos de acción del arsénico inorgánico y de sus metabolitos metilados sobre la mucosa gastrointestinal no son bien conocidos. Estudios recientes in vitro utilizando líneas celulares humanas de epitelio intestinal, han mostrado que las especies trivalentes de As producen diferentes grados de toxicidad, induciendo apoptosis y necrosis celular. Los mecanismos que afectan la viabilidad celular están asociados a la formación de especies reactivas de oxígeno, reducción de glutatión y peroxidación lipídica. Las especies pentavalentes de As no mostraron toxicidad (Calatayud y col. 2013).
2.8. Efectos sobre el sistema inmune
El As juega un rol importante en la disrupción de la inmunidad innata y la inmunidad mediada por células (Selgrade 2007, Dangleben y col. 2013). Trabajos recientes que intentaron dilucidar los mecanismos por los que estos efectos se producen, sugieren que la respuesta a proteínas no plegadas (UPR) podría ser un mecanismo para explicar la afectación de la inmunidad humoral y la celular tanto en función como en número (Banerjee y col. 2009, Srivastava y col. 2013, Hunt y col. 2014).
Se demostró que el As también interfiere con la vía de transducción de señalización de la activación del complejo receptor de células T (TCR) al aumentar los niveles de fosforilación de las quinasas Lck y Fyn (primeras quinasas asociadas al TCR). El incremento en la secreción del factor de estimulación de granulocitos y monocitos (GM-CSF) produciría un estado de inflamación crónica que se asocia con el desarrollo de cáncer. El As reduce la proporción de células T ayudadoras (CD4) en relación con la cantidad de células T citotóxicas (CD8) [CD4/CD8], este parámetro de inmunosupresión también contribuye a la carcinogénesis. Por lo tanto, repetidas y prolongadas exposiciones a As alteran la activación de linfocitos T y producen cambios funcionales del sistema inmune dependientes de la dosis y de los mecanismos de inmunotoxicidad (Soto-Peña y col. 2006, Soto-Peña y Vega 2008, Vega Loyo 2009).
2.9. Efectos sobre la reproducción y el eje adenohipofisario
Si bien se han reportado efectos adversos del As inorgánico sobre la función reproductiva, poco se conoce de sus acciones sobre la liberación hormonal y los mecanismos de acción sobre la fisiología adenohipofisaria. Estudios en animales han mostrado que el As afecta la salud reproductiva en hembras actuando como citotóxico y como disruptor endócrino, restringiendo la función y estructura del útero, alterando los niveles de gonadotrofinas y esteroides (estradiol), produciendo aumento del peso corporal e intolerancia a la glucosa a bajas dosis (Akram y col. 2009, Chattopadhyay y Ghosh 2010, Rodriguez y col. 2016).
En un trabajo reciente se demostró por primera vez que la administración de As inorgánico es capaz de inducir cambios en la expresión del receptor estrogénico (Reα) en células epiteliales y el estroma del útero (Ronchetti 2014). Este estudio también evidenció que el As afecta de manera directa la adenohipófisis tanto in vivo como in vitro. Es capaz de generar estrés oxidativo en la glándula alterando la expresión de genes como HO-1, MT-1 y TRX-1 (codificantes de la hemooxigenasa 1, metalotioneína 1 y tiorredoxina, respectivamente) (Ronchetti 2014). Además, el As disminuye la viabilidad de las células adenohipofisarias como consecuencia de la inducción de apoptosis y disminuye la liberación de prolactina (PRL). Considerando que la PRL cumple un rol fundamental en la fisiología reproductiva, es probable que el efecto sobre la secreción de esta hormona contribuya a las alteraciones reproductivas que se observan por exposición al As (Ronchetti 2014).
3. Mecanismos de acción asociados a carcinogénesis
La Agencia Internacional de Investigación del Cáncer (IARC, por sus siglas en inglés) inclu yó al As inorgánico en el Grupo 1, agentes o grupos de agentes carcinógenos para los seres humanos, debido a su capacidad para causar cáncer de piel, vejiga, hígado y/o pulmón (Jackson y Grainge 1975, Cantor y Lubin 2007, Liu y Waalkes 2008, IARC 2012). Sin embargo, así como ocurre para la mayoría de los efectos tóxicos del As, los mecanismos por los cuales se desarrollan los cánceres aún permanecen sin esclarecerse. La exposición crónica al As se ha vinculado con una variedad de procesos como proliferación celular, recombinación genética, inducción de genes e inducción de proteínas de choque térmico. En seres humanos, se identificaron algunos factores adicionales de riesgo de desarrollar los efectos deletéreos del As. Estos son: metilación aberrante del DNA, estrés oxidativo, hiperhomocisteinemia y deficiente capacidad para metabolizar (metilar) al As (Niedzwiecki y col. 2013, Howe y col. 2014, Niedzwiecki y col. 2014). A continuación se describen algunos de los mecanismos de acción propuestos para carcinogénesis.
3.1. Genotoxicidad
Aunque parece no ser un mutágeno directo, el As es responsable de causar daño al ADN en células de todos los mamíferos (Faita y col. 2013). El daño puede ser a nivel cromosómico y a nivel del ADN. Numerosos trabajos reportan que la exposición crónica al As produce un incremento de aberraciones cromosómicas, entre las que se mencionan: intercambio de cromátides hermanas, deleciones, micronúcleos, aneuploidías, daño oxidativo y rotura de cadenas (Gradecka y col. 2001, Hughes y col. 2011, Faita y col. 2013). Los arsenicales trivalentes son genotóxicos más potentes que los pentavalentes. El mecanismo molecular más relacionado con este daño es la producción de estrés oxidativo (Liu y col. 2001, Faita y col. 2013).
3.2.Alteración de la reparación del ADN
El As interfiere en la reparación del ADN, afectando enzimas de los sistemas de escisión y reparación de nucleótidos (NER) y de bases (BER) (Hughes y col. 2011, Faita y col. 2013, Holcomb y col. 2017). El sistema NER interviene reparando grandes daños en la doble hélice del ADN y el sistema BER interviene reparando el daño provocado por pequeñas roturas de la cadena simple de ADN. Una revisión reciente destaca el rol de este mecanismo en la carcinogénesis producida por el As (Gentry y col. 2010), principalmente a través de la inhibición de la enzima poli (ADP-ribosa) polimerasa-1, que participa en la reparación del daño al ADN de la célula (Huestis y col. 2016, Zhoe y col. 2016).
3.3. Modificación de histonas
La alteración de la metilación, acetilación y ubiquitinación de histonas puede afectar los procesos de reparación del ADN (He y col. 2010). Las enzimas encargadas de la modificación de histonas son proteínas que se caracterizan por presentar una estructura conocida como dedos de zinc. El As se une a esta estructura dedos de zinc inhibiendo la actividad de las enzimas que actúan sobre las histonas (Tam y col. 2017). En otras palabras, la alteración del proceso de modificación de histonas sería otro mecanismo por el cual el As afecta la reparación del ADN (Society of Toxicology 2017).
3.4. Alteración de la metilación del ADN
La alteración de la metilación del ADN produce cambios en la expresión de los genes. Está descripto que el As induce tanto hipo como hipermetilación del ADN. Estos cambios epigenéticos pueden producir activación de oncogenes o silenciamiento de genes supresores de tumores. Uno de los ejemplos más descriptos es la hipermetilación del gen p53, codificante de una proteína supresora de tumores, la cual tiene un rol en la regulación del ciclo celular (Mass y Wang 1997, Chanda y col. 2006, Intarasunanont y col. 2012). La hipermetilación, con el consecuente silenciamiento de genes supresores de tumores, es un evento temprano implicado en el desarrollo de tumores malignos.
Los mecanismos por los cuales el As produce los cambios en la metilación del ADN no están claros. La hipometilación podría deberse a la inhibición de las ADN metiltransferasas, a la competencia (entre metiltransferasas) por la S-adenosil metionina (SAM), involucrada también en el metabolismo del As, o a la depleción de SAM por deficiencias nutricionales (Reichard y Puga 2010, Hughes y col. 2011). La hipermetilación resulta más difícil de explicar.
3.5. Proliferación celular
Una manifestación característica de la exposición crónica al As es la queratosis. En respuesta a estímulos ambientales, los que ratinocitos producen y secretan una serie de citocinas proinflamatorias y quimiocitocinas, como la interleuquina-1α, el factor de necrosis tumoral α, la interleuquina-8, así como factores de crecimiento como el factor de crecimiento transformante α y el factor estimulante de colonias de granulocitos y macrófagos (Germolec y col. 1996, Dodmane y col. 2013). La sobreproducción de estos compuestos se ha vinculado con varios procesos patológicos, incluidas las neoplasias.
3.6. Transducción de señales
La transducción de señales es el mecanismo por el cual una señal del exterior de una célula se transmite al interior y provoca una alteración en la expresión de algunos genes de esa célula. El As puede afectar este mecanismo llevando a la activación o inhibición de factores de transcripción (proteínas que se unen al ADN y regulan la transcripción génica) (Hughes y col. 2011). Entre los factores de trascripción más estudiados se encuentran el AP-1, el NF-κβ y p53 (Flora 2011).
3.7. Co-carcinogénesis
El As puede desempeñar el rol de co-carcinógeno promoviendo el desarrollo de cánceres iniciados por otros agentes físicos o químicos. Muchos trabajos dan cuenta de la capacidad del As de promover el daño cromosómico inducido (iniciado) por la radiación ultravioleta (UV) en células de mamíferos (Hughes 2002, Rossman y col. 2004, Hughes y col. 2011), a través de la inhibición, por parte del As, de los procesos de reparación del daño al ADN (Qin y col. 2008, Hughes y col. 2011, Hunt y col. 2014). El As entonces podría actuar como co-carcinógeno promoviendo el desarrollo de cáncer de piel debido a exposición a radiación UV. El As demostró también actuar incrementando la captación, por parte de la célula, de benzopireno diolepóxido (metabolito del benzo[a]pireno) y, de esta forma, favorecer la formación de aductos de ADN (Shen y col 2006). Este mecanismo explicaría el rol del As como co-carcinógeno, promotor del cáncer de pulmón debido a la exposición a hidrocarburos aromáticos policíclicos (presentes, por ejemplo, en el humo del tabaco).
Conclusiones
El As interfiere en una gran cantidad de procesos biológicos fundamentales para la célula como el almacenamiento y la obtención de energía, la reparación del daño al ADN o la inducción de apoptosis, entre muchos otros. Sin embargo, y a pesar de la cantidad de información disponible acerca de los potenciales mecanismos de acción tóxica, todavía quedan muchos huecos por llenar. Por ejemplo, es posible que los mecanismos involucrados requieran distintas concentraciones de As o distintos tiempos de exposición para manifestarse. También es posible que, aunque se presumen interrelacionados e interdependientes, no todos los mecanismos tengan lugar en todas las células, tejidos y órganos. Por último, muchos de los mecanismos de acción propuestos se estudiaron en modelos experimentales in vitro o en modelos animales lo cual implica realizar extrapolaciones entre dosis, vías de administración, tiempos de exposición y especies animales. En definitiva, la investigación sobre la toxicodinamia del As, si bien deja aún incertezas, ha permitido esclarecer los mecanismos moleculares implicados en algunos de los efectos tóxicos que produce este compuesto en el humano.
Bibliografía citada
1. Akram Z., Jalali S., Shami S.A., Ahmad L., Batool S., Kalsoom O. Adverse effects of arsenic exposure on uterine function and structure in female rat. Exp Toxicol Pathol. 2010; 62(4):451-9. [ Links ]
2. Banerjee N., Banerjee S., Sen R., Bandyopadhyay A., Sarma N., Majumder P., Das J.K., Chatterjee M., Kabir S.N., Giri A.K. Chronic arsenic exposure impairs macrophage functions in the exposed individuals. J Clin Immunol. 2009; 29 (5): 582-94. [ Links ]
3. Calatayud M., Devesa V., Vélez D. Differential toxicity and gene expression in Caco-2 cells exposed to arsenic species. Toxicology Letters. 2013; 218(1): 70-80. [ Links ]
4. Cantor K.P., Lubin J.H. Arsenic, internal cancers, and issues in inference from studies of low-level exposures in human populations. Toxicol Appl Pharmacol. 2007; 222 (3): 252-7. [ Links ]
5. Chanda S., Dasgupta U.B., Guha Mazumder D., Gupta M., Chaudhuri U., Lahiri S., Das S., Ghosh N., Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic- exposed people with and without malignancy. Toxicol Sci. 2006; 89 (2): 431-7. [ Links ]
6. Chattopadhyay S., Ghosh D. The involvement of hypophyseal-gonadal and hypophysealadrenal axes in arsenic-mediated ovarian and uterine toxicity: modulation by hCG. J Biochem Mol Toxicol. 2010; 24(1):29-41. [ Links ]
7. Dangleben N.L., Skibola C.F., Smith M.T. Arsenic immunotoxicity: a review. Environ Health. 2013;12(1):73. [ Links ]
8. Dodmane P.R., Arnold L.L., Kakiuchi-Kiyota S., Qiu F., Liu X., Rennard S.I., Cohen S.M. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology. 2013; 312: 18-29. [ Links ]
9. Edwards D.H., Li Y., Ellinsworth D.C., Griffith T.M. The effect of inorganic arsenic on endothelium- dependent relaxation: Role of NADPH oxidase and hydrogen peroxide. Toxicology. 2013;306:50-58. [ Links ]
10. Ellinsworth D.C. Arsenic, reactive oxygen, and endothelial dysfunction. J Pharmacol Exp Ther. 2015;353 (3): 458-64. [ Links ]
11. Faita F., Cori L., Bianchi F., Andreassi M.G. Arsenic-induced genotoxicity and genetic susceptibility to arsenic-related pathologies. Int J Environ Res Public Health. 2013;10 (4):1527-46. [ Links ]
12. Flora S.J. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2011; 51(2):257-81. [ Links ]
13. Florea A.M., Yamoah E.N., Dopp E. Intracellular calcium disturbances induced by arsenic and its methylated derivatives in relation to genomic damage and apoptosis induction. Environ Health Perspect. 2005;113(6):659-64. [ Links ]
14. Gentry P.R., McDonald T.B., Sullivan D.E., Shipp A.M., Yager J.W., Clewell H.J. 3rd. Analysis of genomic dose-response information on arsenic to inform key events in a mode of action for carcinogenicity. Environ Mol Mutagen. 2010;51(1):1-14. [ Links ]
15. Germolec D.R., Yoshida T., Gaido K., Wilmer J.L., Simeonova P.P., Kayama F., Burleson F., Dong W., Lange R.W., Luster M.I. Arsenic induces overexpression of growth factors in human keratinocytes. Toxicol Appl Pharmacol. 1996;141(1):308-18. [ Links ]
16. Gradecka D., Palus J., Wasowicz W. Selected mechanisms of genotoxic effects of inorganic arsenic compounds. Int J Occup Med Environ Health. 2001;14(4):317-28. [ Links ]
17. He F., Umehara T., Saito K., Harada T., Watanabe S., Yabuki T., Kigawa T., Takahashi M., Kuwasako K., Tsuda K., Matsuda T., Aoki M., Seki E., Kobayashi N., Güntert P., Yokoyama S., Muto Y. Structural insight into the zinc finger CW domain as a histone modification reader. Structure. 2010;18(9):1127-39. [ Links ]
18. Hei T.K., Filipic M. Role of oxidative damage in the genotoxicity of arsenic. Free Radic Biol Med. 2004;37(5):574-81. [ Links ]
19. Holcomb N., Goswami M., Han S.G., Scott T., D'Orazio J., Orren D.K., Gairola C.G., Mellon I. Inorganic arsenic inhibits the nucleotide excision repair pathway and reduces the expression of XPC. DNA Repair (Amst). 2017;52:70-80. [ Links ]
20. Howe C.G., Niedzwiecki M.M., Hall M.N., Liu X., Ilievski V., Slavkovich V., Alam S., Siddique A.B., Graziano J.H., Gamble M.V. Folate and cobalamin modify associations between Sadenosylmethionine and methylated arsenic metabolites in arsenic-exposed Bangladeshi adults. J Nutr. 2014;144(5):690-7. [ Links ]
21. Huestis J., Zhou X., Chen L., Feng C., Hudson L.G., Liu K.J. Kinetics and thermodynamics of zinc(II) and arsenic(III) binding to XPA and PARP-1 zinc finger peptides. J Inorg Biochem. 2016;163:45-52. [ Links ]
22. Hughes M.F. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133(1):1-16. [ Links ]
23. Hughes M.F., Beck B.D., Chen Y., Lewis A.S., Thomas D.J. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123(2):305-32. [ Links ]
24. Hunt K.M., Srivastava R.K., Elmets C.A., Athar M. The mechanistic basis of arsenicosis: pathogenesis of skin cancer. Cancer Lett. 2014;354(2):211-9. [ Links ]
25. Intarasunanont P., Navasumrit P., Waraprasit S., Chaisatra K., Suk W.A., Mahidol C., Ruchirawat M. Effects of arsenic exposure on DNA methylation in cord blood samples from newborn babies and in a human lymphoblast cell line. Environ Health. 2012;11:31. [ Links ]
26. International Agency for Research on Cancer (IARC). Monographs 100C. Arsenic and arsenic compounds. 2012. [en línea]. Disponible en: http://monographs.iarc.fr/ENG/Monographs/vol100C/mono100C-6.pdf (Consulta 24 de noviembre de 2017) [ Links ]
27. Jackson R., Grainge J.W. Arsenic and cancer. Can Med Assoc J. 1975;113(5):396–401.
28. Jomova K., Jenisova Z., Feszterova M., Baros S., Liska J., Hudecova D., Rhodes C.J., Valko M. Arsenic: toxicity, oxidative stress and human disease. J Appl Toxicol. 2011;31:95–107.
29. Kachinskas D.J., Phillips M.A., Qin Q., Stokes J.D., Rice R.H. Arsenate perturbation of human keratinocyte differentiation. Cell Growth Differ. 1994;5(11):1235-41. [ Links ]
30. Li C., Xu J., Li F., Chaudhary S.C., Weng Z., Wen J., Elmets C.A., Ahsan H., Athar M. Unfolded protein response signaling and MAP kinase pathways underlie pathogenesis of arsenic- induced cutaneous inflammation. Cancer Prev Res (Phila). 2011;4(12):2101-9. [ Links ]
31. Li Y., Zhang Y., Gao Y., Zhang W., Cui X., Liu J., Wei Y. Arsenic Induces Thioredoxin 1 and Apoptosis in Human Liver HHL-5 Cells. Biol Trace Elem Res. 2018;181(2):234-241. [ Links ]
32. Liao W.T., Chang K.L., Yu C.L., Chen G.S., Chang L.W., Yu H.S. Arsenic induces human keratinocyte apoptosis by the FAS/FAS ligand pathway, which correlates with alterations in nuclear factor-kappa B and activator protein-1 activity. J Invest Dermatol. 2004;122(1):125-9. [ Links ]
33. Liu J., Waalkes M.P. Liver is a target of arsenic carcinogenesis. Toxicol Sci. 2008;105(1):24-32. [ Links ]
34. Liu S.X., Athar M., Lippai I., Waldren C., Hei T.K. Induction of oxyradicals by arsenic: implication for mechanism of genotoxicity. Proc Natl Acad Sci U S A. 2001;98(4):1643-8. [ Links ]
35. Lu T.H., Tseng T.J., Su C.C., Tang F.C., Yen C.C., Liu Y.Y., Yang C.Y., Wu C.C., Chen K.L., Hung D.Z., Chen Y.W. Arsenic induces reactive oxygen species-caused neuronal cell apoptosis through JNK/ERK-mediated mitochondria- dependent and GRP 78/CHOP-regulated pathways. Toxicol Lett. 2014;224(1):130-40. [ Links ]
36. Mass M.J., Wang L. Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat Res. 1997;386(3):263-77. [ Links ]
37. Matos R.C., Bessa M., Oliveira H., Gonçalves F., de Lourdes Pereira M., Nunes B. Mechanisms of kidney toxicity for chromium- and arsenic-based preservatives: potential involvement of a pro-oxidative pathway. Environ Toxicol Pharmacol. 2013;36(3):929-36. [ Links ]
38. Mazumder D.N. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol Appl Pharmacol. 2005;206 (2):169-75. [ Links ]
39. Niedzwiecki M.M., Hall M.N., Liu X., Oka J., Harper K.N., Slavkovich V., Ilievski V., Levy D., van Geen A., Mey J.L., Alam S., Siddique A.B., Parvez F., Graziano J.H., Gamble M.V. A doseresponse study of arsenic exposure and global methylation of peripheral blood mononuclear cell DNA in Bangladeshi adults. Environ Health Perspect. 2013;121(11-12):1306-12. [ Links ]
40. Niedzwiecki M.M., Hall M.N., Liu X., Slavkovich V., Ilievski V., Levy D., Alam S., Siddique A.B., Parvez F., Graziano J.H., Gamble M.V. Interaction of plasma glutathione redox and folate deficiency on arsenic methylation capacity in Bangladeshi adults. Free Radic Biol Med. 2014;73:67-74. [ Links ]
41. Pi J., Diwan B.A., Suna Y., Liua J., Qua W., Hed Y., Stybloe M., Waalkesa M.P. Arsenicinduced malignant transformation of human keratinocytes: Involvement of Nrf2. Free Radic Biol Med. 2008;45(5):651–658.
42. Prakash C., Soni M., Kumar V. Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. J Appl Toxicol. 2016;36(2):179-88. [ Links ]
43. Qin X.J., Hudson L.G., Liu W., Timmins G.S., Liu K.J. Low concentration of arsenite exacerbates UVR-induced DNA strand breaks by inhibiting PARP-1 activity. Toxicol Appl Pharmacol. 2008;232(1):41-50. [ Links ]
44. Reichard J.F., Puga A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics. 2010;2(1):87-104. [ Links ]
45. Rocha R.A., Gimeno-Alcañiz J.V., Martín-Ibañez R., Canals J.M., Vélez D., Devesa V. Arsenic and fluoride induce neural progenitor cell apoptosis. Toxicol Lett. 2011;203(3):237-44. [ Links ]
46. Rodriguez K.F., Ungewitter E.K., Crespo-Mejias Y., Liu C., Nicol B., Kissling G.E., Yao H.H. Effects of in utero exposure to arsenic during the second half of gestation on reproductive end points and metabolic parameters in female CD-1 mice. Environ Health Perspect. 2016;124:336-343. [ Links ]
47. Rodríguez-Barranco M., Lacasaña M., Aguilar- Garduño C., Alguacil J., Gil F., González- Alzaga B., Rojas-García A. Association of arsenic, cadmium and manganese exposure with neurodevelopment and behavioural disorders in children: a systematic review and meta-analysis. Sci Total Environ. 2013;454-455:562-77. [ Links ]
48. Ronchetti S.A. Efectos del arsénico sobre la adenohipófisis. Mecanismos de acción. Tesis doctoral. Biblioteca Digital FCEN-UBA. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires, 2014. pp 147. www.digital.bl.fcen.uba.ar. [ Links ]
49. Rossman T.G., Uddin A.N., Burns F.J. Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol Appl Pharmacol. 2004;198 (3):394-404. [ Links ]
50. Samikkannu T., Chen C.H., Yih L.H., Wang A.S., Lin S.Y., Chen T.C., Jan K.Y. Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem Res Toxicol. 2003;16(3):409-14. [ Links ]
51. Sánchez-Peña L.C., Petrosyan P., Morales M., González N.B., Gutiérrez-Ospina G., Del Razo L.M., Gonsebatt M.E. Arsenic species, AS3MT amount, and AS3MT gene expression in different brain regions of mouse exposed to arsenite. Environ Res. 2010;110(5):428-34. [ Links ]
52. Santra A., Maiti A., Das S., Lahiri S., Charkaborty S.K., Mazumder D.N. Hepatic damage caused by chronic arsenic toxicity in experimental animals. J Toxicol Clin Toxicol. 2000;38(4):395-405. [ Links ]
53. Sasaki A., Oshima Y., Fujimura A. An approach to elucidate potential mechanism of renal toxicity of arsenic trioxide. Exp Hematol. 2007;35 (2):252-62. [ Links ]
54. Selgrade M.K. Immunotoxicity: the risk is real. Toxicol Sci. 2007;100(2):328-32. [ Links ]
55. Shen S., Lee J., Sun X., Wang H., Weinfeld M., Le X.C. Elevation of cellular BPDE uptake by human cells: a possible factor contributing to co-carcinogenicity by arsenite. Environ Health Perspect. 2006;114(12):1832-7. [ Links ]
56. Shen S., Li X.F., Cullen W.R., Weinfeld M., Le X.C. Arsenic Binding to Proteins. Chem Rev. 2013;113:7769-7792. [ Links ]
57. Singh A.P., Goel R.K., Kaur T. Mechanisms Pertaining to Arsenic Toxicity. Toxicol Int. 2011;18(2):87–93.
58. Society of Toxicology (SOT). The Complexities in Assessing the Risk to Public Health of Low-Level Arsenic Exposure. SOT Issue Statement. [en línea] SOT Council, November 2017. Disponible en: https://www.toxicology.org/pubs/statements/SOT-Low-Level-Arsenic-Exposure-Issue-Statement.pdf [Consulta: 6 de diciembre de 2017] [ Links ]
59. Soto-Peña G.A., Luna A.L., Acosta-Saavedra L., Conde-Moo P., López-Carrillo L., Cebrián M. E., Bastida M., Calderón-Aranda E.S., Vega L. Assessment of lymphocyte subpopulations and cytokine secretion in children exposed to arsenic. The FASEB Journal. 2006;20(6):779-781. [ Links ]
60. Soto-Peña G.A., Vega L. Arsenic interferes with the signaling transduction pathway of T cell receptor activation by increasing basal and induced phosphorylation of Lck and Fyn in spleen cells. Toxicology and Applied Pharmacology. 2008;230(2):216-226. [ Links ]
61. Srivastava R.K., Li C., Chaudhary S.C., Ballestas M.E., Elmets C.A., Robbins D.J., Matalon S., Deshane J.S., Afaq F., Bickers D.R., Athar M. Unfolded protein response (UPR) signaling regulates arsenic trioxide-mediated macrophage innate immune function disruption. Toxicol Appl Pharmacol. 2013;272(3):879-87. [ Links ]
62. Tam L.M., Jiang J., Wang P., Li L., Miao W., Dong X., Wang Y. Arsenite Binds to the Zinc Finger Motif of TIP60 Histone Acetyltransferase and Induces Its Degradation via the 26S Proteasome. Chem Res Toxicol. 2017;30 (9):1685-1693. [ Links ]
63. Vahidnia A., Romijn F., van der Voet G.B., de Wolff F.A. Arsenic-induced neurotoxicity in relation to toxicokinetics: effects on sciatic nerve proteins. Chem Biol Interact. 2008;176 (2-3):188-95. [ Links ]
64. Vahidnia A., van der Voet G.B., de Wolff F.A. Arsenic neurotoxicity--a review. Hum Exp Toxicol. 2007;26(10):823-32. [ Links ]
65. Vega Loyo L. Mecanismos moleculares de los efectos biológicos del arsénico. En: Bustos Jaimes I., Castañeda Patlán C., Rendón Huerta E., Reyes Vivas H., Romero Álvarez I., (eds). Mensaje Bioquímico, Vol XXXIII. Depto de Bioquímica, Fac de Medicina, Universidad Nacional Autónoma de México. Cd Universitaria, México, DF, MÉXICO (2009). http://bq.unam.mx/mensajebioquimico. [ Links ]
66. Zhou X., Cooper K.L., Huestis J., Xu H., Burchiel S.W., Hudson L.G., Liu K.J. S-nitrosation on zinc finger motif of PARP-1 as a mechanism of DNA repair inhibition by arsenite. Oncotarget. 2016;7(49):80482-80492. [ Links ]

