










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkPhyton (Buenos Aires)
versión On-line ISSN 1851-5657
Phyton (B. Aires) vol.79 no.2 Vicente López jul./dic. 2010
ARTÍCULOS ORIGINALES
Metagenómica de suelos: grandes desafíos y nuevas oportunidades biotecnológicas
Soil Metagenomics: new challenges and biotechnological opportunities
Hernández-León1 R, I Velázquez-Sepúlveda1, MC Orozco-Mosqueda1, G Santoyo1
1 Laboratorio de Recombinación y Diversidad Genómica, Instituto de Investigaciones Químico Biológicas, Universidad Michoacana de San Nicolás de Hidalgo. Ciudad Universitaria, Edificio A1´, Morelia, Michoacán, México. C.P. 58030.
Address Correspondence to: Dr. Gustavo Santoyo, e-mail: gustavo_santoyo@yahoo.com; gsantoyo@umich.mx
Recibido - Received 21.V.2010.
Aceptado - Accepted 17.VIII.2010.
Resumen. El suelo es un sistema complejo que alberga una gran cantidad y diversidad de microorganismos. Hasta hace poco, sólo se podía tener acceso al estudio de un pequeño porcentaje de la microbiota que habita en este ecosistema. Actualmente, con la metagenómica se ha logrado conocer y estudiar en más detalle todo ese material genómico desconocido. Se han descubierto nuevas moléculas con aplicaciones biotecnológicas y se ha profundizado en entender mejor las diferentes interacciones microbiológicas en diversos ambientes, algunos con características extremas. En este ensayo se analizaron los trabajos más recientes en el área de la metagenómica, con énfasis en aquellos relacionados con el suelo. Asimismo, se describen los logros alcanzados por el proyecto Metacontrol (metagenómica de suelos supresores de enfermedades), y finalmente, proponemos los diferentes desafíos que ha de superar la metagenómica, así como las nuevas oportunidades biotecnológicas que surgen con esta nueva ciencia.
Palabras clave: Metagenómica; Suelo; Microorganismos.
Abstract. Soil is a complex system that includes a great number and diversity of microorganisms. Until recently, only a small percentage of the bioma was known and could be studied. Currently, it is possible to have a deeper knowledge of all that unknown genomic material with the development of new tools, like metagenomics. New molecules have been discovered with various biotechnological applications, and knowledge of the diverse microbiological interactions in several environments, some of them with extreme life conditions, is much higher. We analyze the most recent literature in the field of metagenomics in this study, especially that related with the soil. Achievements of the Metacontrol project (metagenomics of diseasesuppressive soils) are also described. We finally propose various challenges to be overcome in metagenomics, and new biotechnological opportunities emerging with this new science.
Keywords: Metagenomics; Soil; Microorganisms.
Introducción
La metagenómica es una ciencia que surge como una rama de las ciencias genómicas, la cual se refiere al estudio del metagenoma de un nicho en particular (Handelsman, 2004; Riesenfeld et al., 2004). El metagenoma se puede definir como el total de ADN de una muestra ambiental. Hasta el momento, se han investigado metagenomas de diversos ambientes, incluyendo ecosistemas acuáticos, minas, suelos agrícolas y forestales, entre otros (Rondon et al., 2000; Wang et al., 2000; Lee et al., 2004; Tyson et al., 2004; Craig et al., 2009). Lo relevante de estos estudios es que cada uno de ellos ha mostrado diferentes aspectos para estudiar y analizar. En algunos casos, se han descubierto novedosos elementos genéticos que podrían tener aplicación en la industria (Rondon et al., 2000; Wang et al., 2009), mientras que en otros, han aportado novedosos aspectos de la ecología microbiana en un ecosistema en particular (Tyson et al., 2004).
Mediante técnicas convencionales de cultivo en laboratorio se ha estudiado la diversidad microbiana y sus metabolitos. En particular, se ha propuesto que entre 80-90% de los microorganismos que habitan el suelo son desconocidos (Alexander 1977). Esto representa una limitante para descubrir el verdadero potencial genético de estos sistemas. Al estudiar el metagenoma de un ambiente en particular, es muy probable que este consista en gran parte de bacterias u otros organismos no cultivables; sólo conocemos una pequeña proporción de éstos, los cuales podemos reproducir en condiciones de laboratorio (Handelsman 2004; Riesenfeld et al., 2004). De esta manera, el ADN metagenómico se puede clonar en vectores y expresar en diversos huéspedes procariotes, dependiendo de los objetivos del estudio (Rondon et al., 2000; Wang et al., 2000). Todo este material genético podría codificar nuevas o mejores actividades metabólicas. De la misma manera, se pueden emplear métodos de secuenciación masiva que generarán genomas completos de organismos no cultivables (Handelsman, 2004; Tyson et al., 2004).
Metagenómica de la diversidad microbiana no cultivable
Los microorganismos son responsables de una parte importante de los ciclos biogeoquímicos, y por lo tanto, influyen significativamente en la vida terrestre. Sin embargo, nuestro conocimiento de la vida microbiana, y el papel que ésta juega en el ambiente, es todavía poco entendido. Más difícil aún es tratar de conocer la diversidad microbiana; por ejemplo, se ha estimado que existen alrededor de 2x106 especies bacterianas en al ambiente marino, mientras que una muestra de suelo podría contener hasta 4x106 diferentes taxa (Curtis et al., 2002). Esto indica que su estudio es un gran desafío de la microbiología actual.
Un problema obvio al que nos enfrentamos para conocer la diversidad microbiana, y en particular las bacterias, es que estas son invisibles para el ojo humano, por lo que se han desarrollado métodos para cultivarlas en laboratorio (Handelsman, 2004).
Desafortunadamente, se estima que sólo un pequeño porcentaje (0,1 - 10%) de las bacterias son cultivables (Rondon, 1999). Esto implica que la gran mayoría de las bacterias son no cultivables, y por lo tanto, desconocidas para el ser humano. Una probable explicación es el desconocimiento de sus requerimientos nutricionales y fisiológicos para su crecimiento. Este hecho ha limitado enormemente nuestro conocimiento sobre la diversidad bacteriana. Para contrarrestar esta limitante, se han desarrollado métodos para poder aislar y amplificar el material genético de bacterias no cultivables en diferentes ambientes.
Una de las técnicas más usadas desde hace algunas décadas es la amplificación de los genes ribosomales que codifican para la subunidad 16S (ADNr 16S). Este marcador es una poderosa herramienta que ha sido ampliamente usada en clasificaciones filogenéticas, debido a que su secuencia es altamente conservada (Woese y Fox, 1977; Pace et al., 1986; Woese, 1987). La amplificación de los ADNr 16S se realiza por medio de la reacción en cadena de la polimerasa o PCR, por sus siglas en inglés, empleando oligonucleótidos o primers específicos. Esto se realiza para amplificar los ADNr 16S de bacterias u otros grupos como arqueobacterias. En el caso de organismos eucariontes, se emplean las secuencias ADNr 18S (Baker et al., 2003). Los 16S posteriormente se pueden clonar en vectores o plásmidos. Dichos vectores son transferidos a bacterias huésped como Escherichia coli, para crear bibliotecas o bancos de clonas, conteniendo las secuencias ADNr 16S de diversos microorganismos de forma separada. Esto permite el análisis individual de cada una de las secuencias clonadas utilizando diversos métodos, tales como secuenciación, restricción de los 16S ribosomales con nucleasas (ARDRA), etc. (Escalante- Lozada et al., 2004).
De esta manera, miles de secuencias de ADNr 16S de diversos microorganismos, cultivables y no cultivables, son reportadas a las bases de datos como el GenBank del National Center for Biotechnology Information (NCBI), y el Ribosomal Database Project (RDP), cuya información crece continuamente. Asimismo, se ha informado el descubrimiento de nuevos grupos taxonómicos a través de los ADNr 16S usando la amplificación de secuencias de organismos no cultivables obtenidas de ADN ambiental (Hugenholtz y Pace, 1996; Huber et al., 2002). Actualmente, un gran porcentaje de los genes 16S eubacterianos que se han reportado, corresponden a bacterias que no se han podido cultivar en laboratorio. La situación es más drástica en el caso de arqueobacterias, debido a que se las puede encontrar en ambientes extremos, con altas temperaturas, bajos pHs, metales pesados, concentraciones salinas, etc. Incluso, la división Korarqueota (o Korarchaeota) contiene únicamente secuencias ribosomales sin alguna especie cultivable hasta el momento (Birtrim et al., 1997; Garrett y Klenk, 2007). Sin embargo, cabe destacar que existe un gran esfuerzo internacional por secuenciar genomas completos de organismos no cultivables. Esto aportará un mayor conocimiento de su fisiología y probablemente se podrán establecer condiciones en laboratorio para su cultivo.
Estrategias y métodos para la construcción de bibliotecas metagenómicas
La metagenómica estudia el total del ADN que podemos encontrar en un nicho específico. Para acceder a este metagenoma se han desarrollado diferentes metodologías y vías para su análisis. En la Figura 1 se muestra un ejemplo de un estudio metagenómico con una estrategia general y siguiendo los pasos básicos en la investigación: (I) Se colecta la muestra de interés; (II) se aísla el ADN metagenómico (en estudios de metagenómica funcional se puede aislar ARN); (III) el metagenoma puede tomar tres vías: a) aislamiento por PCR de los 16S para conocer la diversidad bacteriana (o de otras divisiones como arqueas o eucariontes) del metagenoma, b) digestión y clonación en vectores de expresión, y/o c) secuenciación directa de la muestra. En el paso final, (IV) se analizan las clonas en busca de actividades de interés. Estos son los pasos esenciales para la construcción de bibliotecas metagenómicas, lo cual puede resultar rutinario en algunos laboratorios; sin embargo, el aislamiento de ADN metagenómico de algunos suelos, con una pureza suficiente como para ser usado como templado en reacciones de PCR, digestión y clonación, puede ser complicado. Algunos suelos pueden contener una gran cantidad de ácidos húmicos o metales pesados, los cuales pueden inhibir la actividad de ADN polimerasas o nucleasas (Tsai et al., 1992). Debido a esto, se recomienda emplear diversos protocolos o kits comerciales específicos para cada tipo de suelo que mejoren la pureza del ADN metagenómico, así como el uso de ADN polimerasas modificadas que pueden polimerizar reacciones en presencia de compuestos inhibidores (Kermekchiev et al., 2009).
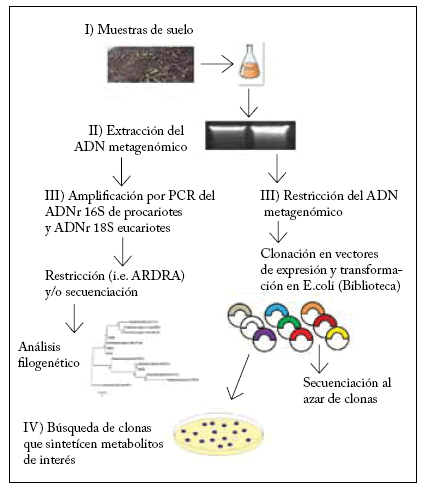
Fig. 1. Protocolo general de análisis del metagenoma de una muestra de suelo. En un primer paso se aísla el suelo que se desea estudiar (I) y se extrae el ADN metagenómico (II). Posteriormente se puede analizar la diversidad microbiana a través de la amplificación de los genes ribosomales 16S y/o llevar a cabo la restricción del ADN, así como su clonación en vectores (III). Finalmente se realiza la búsqueda de funciones o actividades enzimáticas (IV).
Fig. 1. Diagram showing the metagenomics analysis of a soil sample. (I) Soil isolation for study. (II) Extraction of metagenomics DNA. (III) Analysis of the microbial density through amplification of ribosomal genes 16S and/or DNA restriction, or its cloning in vectors. (IV) Search for enzymatic functions or activities.
Metagenómica de diversos ambientes
El conocimiento del metagenoma de un ambiente en particular puede permitirnos conocer diversos aspectos de la vida microbiana que allí se está generando, genes que se expresan bajo esas condiciones ambientales, o quizás, las diferentes interacciones ecológicas de ese nicho particular. Persiguiendo estos objetivos se ha investigado el metagenoma de ambientes muy diversos. Por ejemplo, Venter y colaboradores (2004) secuenciaron más de un billón de pares de bases de ADN del mar Sargasso. Los autores reportan el descubrimiento de alrededor de 1800 especies genómicas; 1,2 millones de nuevos genes y más de 700 genes nuevos tipo rodopsina (Venter et al., 2004). Martín-Cuadrado y colegas (2007) también publicaron otro trabajo sobre metagenómica de ambientes marinos. Ellos aislaron el metagenoma y construyeron una biblioteca del mar Mediterráneo a 3.000 m de profundidad. Asimismo, analizaron la diversidad bacteriana por medio de la secuenciación de los 16S, encontrando que el grupo más abundante fue el de rhizobiales, dentro de las alfaproteobacterias, seguido de bacterias gram-positivas, actinobacterias y firmicutes. Del grupo de las arqueas, el genoma de Cenarchaeum symbiosum fue el que presentó mayor proporción. Algunos otros planctomycetes fueron también reportados, tales como Blastopirellula marina y Rhodopirellula báltica, los cuales son organismos comunes de océanos oligotróficos. En el análisis de la biblioteca metagenómica reportaron la presencia de altos porcentajes de genes que codifican para deshidrogenasas, tales como algunos genes cox. Esto sugiere que la oxidación del monóxido de carbono podría ser una fuente importante de energía en aguas marinas profundas (Martín-Cuadrado et al., 2007).
Por medio de la metagenómica también es posible la reconstrucción de genomas completos a partir de organismos no cultivables o desconocidos. Tal es el caso del estudio realizado en la Mina Richmond de California, donde se encontró una limitada diversidad microbiana, principalmente por los géneros bacterianos Leptospirillum, Sulfobacillus y Acidomicrobium, además de una especie bacteriana, Ferroplasma acidarmanus. Cabe destacar que el pH de la mina era de 0 a 1, contenía altos niveles de Fe, Zn, Cu y As, el agua tenía poco oxígeno, y su temperatura era de 42 °C. Adicionalmente las únicas fuentes de carbono y nitrógeno sólo fueron en forma de gases. Estas condiciones ambientales son extremas para cualquier tipo de vida microbiana. Sin embargo, son perfectas e interesantes para estudios metagenómicos, debido a que la limitada diversidad bacteriana permitió la reconstrucción casi total de los genomas de Leptospirillum grupo II y Ferroplasma tipo II. En consecuencia, se reportaron diversos genes que codificaban para resistencia a metales pesados, bombas de expulsión de protones, etc. Esta información muestra la adaptación que han tenido estos organismos a ambientes extremos, siendo la metagenómica muy importante para acceder al conocimiento de estos genomas no cultivables en laboratorio (Tyson et al., 2004).
Existen otros estudios metagenómicos: (1) la secuenciación completa de una bacteria del género Buchnera, un endosimbionte de áfidos (Pérez-Brocal et al., 2006), (2) metagenómica del intestino (Gill et al., 2006) y dentadura de humanos (Rudney et al., 2010), etc. Mediante la metagenómica se pudo reconstruir en gran parte el genoma del mamut, una especie extinta (Poinar et al., 2006). Esto nos muestra el potencial que tienen los estudios metagenómicos, y que pronto veremos más publicaciones que generarán un mayor conocimiento de los más diversos ambientes, incluyendo aquellos relacionados con el suelo.
Metagenómica de suelos
El suelo es probablemente uno de los ambientes más complejos por su diversidad y complejidad microbiológica (Daniel, 2005; Mocalli y Benedetti, 2010). Entender la ecología de los microorganismos es otro desafío para la biología, debido a la gran cantidad de interacciones con factores bióticos y abióticos. Es también una excelente oportunidad para lograr un mayor entendimiento de los aspectos evolutivos y biológicos de los microorganismos, así como de sus aspectos ecológicos. Desde el punto de vista de la biotecnología, este ambiente es visto como una gran reserva de enzimas, antibióticos y otros productos naturales por descubrir (Handelsman 2004; Riesefield et al., 2004a; Mori et al., 2008). De hecho, una gran parte de drogas contra el cáncer han sido descubiertas a partir de microorganismos del suelo (Pettit, 2004): por ejemplo, la bleomicina y actinomicina D, que fueron aisladas de Streptomyces verticillius y Actinomyces spp., respectivamente.
Con las herramientas que provee la metagenómica se pretende acelerar el descubrimiento de nuevos compuestos con diversas actividades. En la Tabla 1 se muestran algunos trabajos relevantes de la metagenómica de suelos. Uno de los primeros trabajos sobre metagenómica de suelos fue realizado por Henne y colaboradores (1999). Estos autores construyeron una biblioteca basada en plásmidos con más de 900.000 clonas, buscando aquellas que crecieran en 4-hydroxybutirato como única fuente de energía y carbono. Así, encontraron cinco clonas positivas y con fenotipo estable para utilizar 4-hydroxybutirato. Análisis de la secuencia de los plásmidos aislados resultaron en genes que codificaron para 4-hydroxybutirato deshidrogenasas. Sorprendentemente, algunas secuencias no mostraron similitud con aquellas depositadas en el NCBI (National Center for Biotechnology Information). Esto demuestra que pueden encontrarse nuevas secuencias con funciones conocidas en bibliotecas metagenómicas.
Tabla 1. Trabajos relevantes sobre actividades biológicas encontradas en bibliotecas metagenómicas de suelos.
Table 1. Major research works on biological activities found in metagenomics soil libraries.

Una desventaja de utilizar plásmidos que puedan contener únicamente secuencias pequeñas de ADN (2-10 Kb) muestra la limitante de no poder clonar operones o rutas biosintéticas completas. Para ello, se utilizan vectores tipo BAC (Bacterial Artificial Chromosomes), los cuales pueden llevar clonado hasta 750 kb. Sin embargo, generalmente se trabaja en el orden de 100 a 300 kb (Handelsman, 2004). Los fásmidos (o Fosmid, por sus siglas en inglés), que pueden llevar entre 30 y 40 kb, son otro tipo de vectores con mayor capacidad para llevar ADN de mayor tamaño que los plásmidos. El primer reporte de bibliotecas metagenómicas del suelo, la cual contenía grandes fragmentos de ADN, fue realizado por Rondon y colaboradores (2000). Las bibliotecas están basadas en el vector pBeloBAC11 (Kim et al., 1996), una con más 3.500 clonas y la otra con 24.000 clonas; el tamaño promedio de los insertos fue de 27 kb, aunque algunos fueron de más de 80 kb. Entre las clonas se encontraron actividades de lipasa, amilasa y nucleasa. De las bibliotecas encontraron genes 16S pertenecientes a diversos taxa, incluyendo gram positivas, Proteobacteria, Acidobacterium y Cytophagales. Posteriormente, se informó que unas clonas de esa misma biblioteca presentaban la síntesis de los antibióticos Turbomicina A y B. Dichos compuestos presentaron actividad inhibitoria contra diversas bacterias gram-negativas y gram-positivas, así como contra C. guilliermondii Y001, pero no contra P. ultimum 1033 y F. solani (Rondon et al., 2000; Rondon et al., 2004). En un trabajo reciente, Hernández-León y colaboradores analizaron el metagenoma de suelo rizosférico de plantas de trigo mediante la amplificación y secuenciación de los genes ribosomales 16S. Estos autores encontraron una gran diversidad de bacterias, predominando las gammaproteobacterias. Adicionalmente, se construyó una biblioteca metagenómica y se encontraron actividades hemolíticas y genes de resistencia a diversos antibióticos (Hernández-León y Santoyo, resultados no publicados).
En la metagenómica, la bacteria Escherichia coli es un huésped idóneo para la expresión heteróloga de ADN por su versatilidad genética y otros factores. Sin embargo, también es una limitante para la expresión de algún tipo de proteína que requiera de ciertos factores metabólicos o genéticos, o que simplemente sean tóxicos para E. coli (Wang et al., 2000). Algunos trabajos muestran que genes que producen ciertos compuestos naturales (que no pueden ser sintetizados en E. coli) pueden ser producidos en otros huéspedes, tales como la bacteria Ralstonia metallidurans (Craig et al., 2009). Por tal motivo, se han construido bibliotecas metagenómicas en huéspedes diferentes a E. coli, tales como Streptomyces spp. y Ralstonia metallidurans (Wang et al., 2000; Craig et al., 2009). Conforme se vayan generando mayores datos metagenómicos, se podrá disponer de nuevos huéspedes que permitan la expresión de una mayor diversidad de elementos genéticos.
Metagenómica de suelos donde se suprimen enfermedades
Un suelo supresor se define como aquel donde ciertas enfermedades vegetales no progresan, aún cuando el fitopatógeno está presente (Van Elsas et al., 2008). Esto es debido en gran parte a que los microorganismos benéficos (en dichos suelos) son los responsables de inhibir dichas enfermedades (Steinberg, 2006; Van Elsas et al., 2008). La gran diversidad de compuestos inhibitorios o antibióticos sintetizados en estos suelos juega un papel fundamental en su capacidad de inhibir enfermedades. Por tal motivo, se ha propuesto y desarrollado recientemente un proyecto internacional para explorar dichos suelos en diversas regiones del planeta, en busca de nuevos antibióticos y otros compuestos antimicrobianos, con un enfoque metagenómico. Actualmente el proyecto se conoce como METACONTROL Project (Van Elsas et al., 2008) que se inició en el año 2002 como colaboración entre laboratorios, principalmente de Europa. Hasta el momento se pueden mencionar algunos resultados (Tabla 2); se han informado diversas moléculas que muestran actividades antagonistas hacia diversos patógenos, tales como Rhizoctonia solani, Fusarium sp. y Pythium ultimum (Courtois et al., 2003; Ginolhac et al., 2004; Van Elsas, 2010). Diferentes clonas de bibliotecas metagenómicas construidas se han analizado por secuenciación, y han mostrado similitud a genes poliquetido sintetazas y quitinasas. Algunos de estos genes codifican para nuevos antibióticos poliquetidos (Van Elsas et al., 2008). Es de esperar que en años siguientes se den mayores resultados en este tipo de megaproyectos, donde de manera importante convergen diversas instituciones en un mismo objetivo: desarrollar nuevos métodos de biocontrol de fitopatógenos a través de la metagenómica, aprovechando la diversidad microbiana de suelos donde se suprimen enfermedades.
Tabla 2. Actividades antagónicas informadas del proyecto METACONTROL hasta el 2008 (modificado de van Elsas et al., 2008).
Table 2. Antagonistic activities reported from the project METACONTROL until 2008 (modified from van Elsas et al., 2008).

Conclusiones: desafíos y oportunidades
El suelo es uno de los ambientes donde se presentan muchos desafíos, que lo hacen al mismo tiempo interesante para ser estudiado. Un solo gramo de suelo puede contener miles de especies de organismos procariontes, la mayoría de los cuales reside en la superficie terrestre. De hecho, la mayoría de la biomasa que reside en nuestro planeta es microbiana (Daniel, 2005). Sin embargo, existen aún diversos desafíos que vencer (en la metagenómica y otras áreas relacionadas) para poder acceder a toda esa diversidad microbiana, conocer sus genomas, sus relaciones filogenéticas o sus capacidades metabólicas. En una revisión reciente, Chistoserdova (2010) analizó las diferentes técnicas y novedosas ramas de la metagenómica, como la metatranscriptómica y metaproteómica. Éstas se consideran, junto con las nuevas técnicas de secuenciación, la siguiente generación de tecnologías que acelerarán el descubrimiento de nuevos compuestos. A continuación proponemos algunos desafíos que aún deben superarse: (1) se necesita mejorar los métodos de análisis (¨screening¨) de nuevas o mejores funciones enzimáticas o síntesis de metabolitos, antibióticos. Esto ayudará a analizar miles de clonas en poco tiempo, ahorrando costos y esfuerzo; (2) se requiere desarrollar nuevos vectores de expresión con alta capacidad para clonar fragmentos grandes de ADN (>50 kb), y que además, sean replicables en bacterias gram-positivas (ej. Bacillus sp) y gram-negativas (ej. Escherichia coli). Esto ayudará a la expresión de genes en huéspedes diferentes, ampliando la posibilidad de selección de las funciones de interés; (3) los métodos de secuenciación han mejorado bastante y son cada vez más eficientes; sin embargo, los costos son aún muy altos (en especial los de pirosecuenciación) para la mayoría de los laboratorios en países en desarrollo; (4) se requiere construir bibliotecas empleando huéspedes distintos a E. coli, que no sólo sean huéspedes procariontes sino que incluyan otros grupos como arqueas o eucariontes (Saccharomyces cerevisiae, por ejemplo); (5) se requiere de estudiantes e investigadores en el área de la metagenómica, en especial en países de Latinoamérica, ya que esto permitiría la colaboración multidisciplinaria entre colegas para competir con proyectos internacionales.
Podemos concluir diciendo que la metagenómica es una ciencia relativamente nueva, que está ayudando a entender cómo los microorganismos, cultivables o desconocidos, se adaptan e interactúan con factores bióticos y abióticos en el suelo. A la vez, la metagenómica promete revelar nuevas moléculas, las cuales pueden mejorar diversas aplicaciones biotecnológicas.
AGRADECIMIENTOS
Agradecemos a los revisores por las sugerencias que en gran parte han mejorado el artículo. También a la CIC-UMSNH (Proyectos 2009-2010) por financiar los proyectos en el laboratorio. I.V.-S.y R.H-L. son becarias de maestría del Conacyt-México.
REFERENCIAS
1. Alexander, M. (1977). Introduction to soil microbiology. John Wiley & Sons, New York. p. 472. [ Links ]
2. Baker, G.C., J.J. Smith y D.A. Cowan (2003). Review and re-analysis of domain-specific 16S primers. Journal of Microbiological Methods 55: 541-555. [ Links ]
3. Birtrim, S.B., T.J. Donohue, J. Handelsman, G.P. Roberts y R.M. Goodman (1997). Molecular phylogeny of archaea from soil. Proceedings of the National Academy of Sciences 94: 277-282. [ Links ]
4. Chistoserdova, L. (2010). Recent progress and new challenges in metagenomics for biotechnology. Biotechnol Letters En prensa. [ Links ]
5. Courtois, S., C.M. Cappellano, M. Ball, F.X. Francou, P. Normand, G. Helynck, A. Martinez, S. J. Kolvek, J. Hopke, M.S. Osburne, P.R. August, R. Nalin, M. Guérineau, P. Jeannin, P. Simonet y J.L. Pernodet (2003). Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Applied and Environmental Microbiology 69: 49-55. [ Links ]
6. Craig, J.W., F.Y. Chang y S.F. Brady (2009). Natural products from environmental DNA hosted in Ralstonia metallidurans. ACS Chemical Biology 4: 23-28. [ Links ]
7. Curtis, T.P., W.T. Sloan y J.W. Scannell (2002). Estimating prokaryotic diversity and its limits. Proceedings of the National Academy of Sciences 99: 10494-10499. [ Links ]
8. Daniel, R. (2005). The metagenomics of soil. Nature Review Microbiology 3: 470-478. [ Links ]
9. Escalante-Lozada, A., G. Gosset-Lagarda, A. Martínez-Jiménez y F. Bolívar-Zapata (2004). Diversidad bacteriana del suelo: métodos de estudio no dependientes del cultivo microbiano e implicaciones biotecnológicas. Agrociencia 38: 583-592. [ Links ]
10. Garrett, R.A.y H.P. Klenk (2007). Archaea: evolution, physiology and molecular biology. Blackwell Publishing. p. 388. [ Links ]
11. Gill, S.R., M. Pop, R.T. DeBoy, P.B. Eckburg, P.J. Turnbaugh, B.S. Samuel, J.I. Gordon, D.A. Relman, C.M. Fraser-Liggett y K.E. Nelson (2006). Metagenomic analysis of the human distal gut microbiome. Science 312: 1355-1359. [ Links ]
12. Ginolhac, A., C. Jarrin, B. Gillet, P. Robe, P. Pujic, K. Tuphile, H. Bertrand, T.M. Vogel, G. Perrière, P. Simonet y R. Nalin (2004). Phylogenetic analysis of polyketide synthase Idomains from soil metagenomic libraries allows selection of promising clones. Applied and Environmental Microbiology 70: 5522-5527. [ Links ]
13. Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiology and Molecular Biology Reviews 68: 669-685. [ Links ]
14. Henne, A., R. Daniel, R.A. Schmitz y G. Gottschalk (1999). Construction of environmental DNA libraries in Escherichia coli and screening for the presence of genes conferring utilization of 4-hydroxybutyrate. Applied and Environmental Microbiology 65: 3901-3907. [ Links ]
15. Huber, J.A., D.A. Butterfield y J.A. Baross (2002). Temporal changes in archaeal diversity and chemistry in a mid-ocean ridge subseafloor habitat. Applied and Environmental Microbiology 68: 1585-1594. [ Links ]
16. Hugenholtz, P. y N.R. Pace (1996). Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends in Biotechnology 14: 190-197. [ Links ]
17. Kermekchiev, M.B., L.I. Kirilova, E.E. Vail y W.M. Barnes (2009). Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Research 37:e40. [ Links ]
18. Kim, U.-J., B. B. Birren, T. Slepak, V. Mancino, C. Boysen, H.-L. Kang, M. I. Simon y H. Shizuya (1996). Construction and characterization of a human bacterial artificial chromosome library. Genomics 34: 213-218. [ Links ]
19. Lee SW, K. Won, H.K. Lim, J.C. Kim, G.J. Choi, y K.Y. Cho (2004). Screening for novel lipolytic enzymes from uncultured soil microorganisms. Applied Microbiology and Biotechnology 65: 720-726. [ Links ]
20. Martín-Cuadrado, A.B., P. López-García, J.C. Alba, D. Moreira, L. Monticelli, A. Strittmatter, G. Gottschalk, F. Rodríguez-Valera (2007). Metagenomics of the deep Mediterranean, a warm bathypelagic habitat. PLoS One 2:e914. [ Links ]
21. Mocali, S. y A. Benedetti (2010). Exploring research frontiers in microbiology: the challenge of metagenomics in soil microbiology. Research in Microbiology 161: 497-505. [ Links ]
22. Mori, T, S. Misuza, H. Suenaga y K. Miyazaki (2008). Metagenomic screening for bleomycin resistance genes. Applied and Environmental Microbiology 74: 6803-6805. [ Links ]
23. Pace, N.R., D.A. Stahl, D.J. Lane y G.J. Olsen (1986). The analysis of natural microbial populations by ribosomal RNA sequences. Advances in Microbial Ecology 9: 1-55. [ Links ]
24. Pettit, R.K. (2004). Soil DNA libraries for anticancer drug discovery. Cancer Chemotherapy and Pharmacology 54: 1-6. [ Links ]
25. Poinar, H.N., C. Schwarz, Ji Qi, B. Shapiro, R.D.E. MacPhee, B. Buigues, A. Tikhonov, D.H. Huson, L.P. Tomsho, A. Auch, M. Rampp, W. Miller, S.C. Schuster (2006). Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA. Science 311: 392-394. [ Links ]
26. Pérez-Brocal, V., R. Gil, S. Ramos, A. Lamelas, M. Postigo, J.M. Michelena, F.J. Silva, A. Moya, A. Latorre (2006). A small microbial genome: the end of a long symbiotic relationship? Science 314:312-313. [ Links ]
27. Riesenfeld, C.S., P.D. Schloss y J. Handelsman (2004). Metagenomics: Genomic Analysis of Microbial Communities. Annual Review of Genetics 38: 525-552. [ Links ]
28. Riesenfeld, C.S., M.R. Goodman y J. Handelsman (2004a). Uncultured soil bacteria are a reservoir of new antibiotic resistance genes. Environmental Microbiology 6: 981-989. [ Links ]
29. Rondon, M., M. Goodman y J. Handelsman (1999). The earth's bounty: assessing and accessing the soil microbial diversity. Trends in Biotechnology 17: 403-409. [ Links ]
30. Rondon, M.R., P.R. August, A.D. Bettermann, S.F. Brady, T.H. Grossman, M.R. Liles, K.A. Loiacono, B.A. Lynch, I.A. Mc Neil, C. Minor, Ch.L. Tiong, M. Gilman, M.S. Osburne, J. Clardy, J. Handelsman y R.M. Goodman (2000). Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Applied and Environmental Microbiology 66: 2541-2547. [ Links ]
31. Rudney, J.D., H. Xie, N.L. Rhodus, F.G. Ondrey y T.J. Griffin. (2010). A metaproteomic analysis of the human salivary microbiota by three-dimensional peptide fractionation and tandem mass spectrometry. Molecular Oral Microbiology 25: 38-49. [ Links ]
32. Steinberg, C. (2006). Soil suppressiveness to plant diseases. En: J.D. van Elsas et al., (eds.) Modern Soil Microbiology II. pp. 455-478, CRC Press. [ Links ]
33. Tsai, Y.L. y B.H. Olson (1992). Rapid method for separation of bacterial DNA from humic substances in sediments for polymerase chain reaction. Applied and Environmental Microbiology 58: 2292-2295. [ Links ]
34. Tyson, G.W., J. Chapman, P. Hugenholtz, E.E. Allen, R.J. Ram, P.M. Richardson, V.V. Solovyev, E.M. Rubin, D.S. Rokhsar y J.F. Banfield (2004). Community structure. Nature 428: 37-43. [ Links ]
35. van Elsas, J.D., R. Costa, J. Jansson, S. Sjöling, M. Bailey, R. Nalin, T.M. Vogel y L. van Overbeek (2008). The metagenomics of disease-suppressive soils - experiences from the METACONTROL project. Trends in Biotechnology 26: 591-601. [ Links ]
36. van Elsas, J.D. (2010). A novel protocol for the metagenomic analysis of suppresive soil. Journal of Microbiological Methods En prensa. [ Links ]
37. Venter, J.C., K. Remington, J.F. Heidelberg, A.L. Halern, D. Rusch, J.A. Eisen, D. Wu, I Paulsen, K.E. Nelson, W. Nelson, D.E. Fouts, S. Levy, A.H. Knap, M.W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tilson, C. Pfannkoch, Y. Rogers y H.O. Smith (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304: 66-74. [ Links ]
38. Wang, G.Y., E. Graziani, B. Waters, W. Pan, X. Li, J. McDermott,G. Meurer, G. Saxena, R.J. Andersen y J. Davies (2000). Novel natural products from soil DNA libraries in a streptomycete host. Organic Letters 2: 2401-2404. [ Links ]
39. Watson, R.J. y B. Blackwell (2000). Purification and characterization of a common soil component which inhibits the polymerase chain reaction. Canadian Journal of Microbiology 46: 633-642. [ Links ]
40. Woese, C.R. (1987). Bacterial evolution. Microbiological Reviews 51: 221-271. [ Links ]
41. Woese, C.R. y G.E. Fox (1977). The concept of cellular evolution. Journal of Molecular Evolution 10: 1-6. [ Links ]

