










Servicios Personalizados
Revista

Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO  uBio
uBio
Compartir

 Permalink
PermalinkPhyton (Buenos Aires)
versión On-line ISSN 1851-5657
Phyton (B. Aires) vol.80 no.2 Vicente López jul./dic. 2011
NOTAS DE INVESTIGACIÓN
Microbial diversity, metagenomics and the Yucatán aquifer
Diversidad microbiana, metagenómica y el acuífero de Yucatán
Rojas-Herrera R1, M Zamudio-Maya1, L Arena-Ortiz2, RC Pless3, A O'Connor-Sánchez4
1 Facultad de Ingeniería Química. Universidad Autónoma de Yucatán. Periférico Nte. Km. 33.5, Tablaje Catastral 13615, Col. Chuburná de Hidalgo Inn, Mérida, Yucatán, México. C.P. 97203. Rafael Rojas-Herrera, e-mail: r.rojas@uady.mx; Marcela Zamudio-Maya, e-mail: marcela.zamudio@uady.mx
2 Unidad Multidisciplinaria de Docencia e Investigación-Sisal. Facultad de Ciencias, Universidad Nacional Autónoma de México. Sisal, Yucatán, México. Leticia Arena-Ortiz, e-mail: arena.leticia@gmail.com
3 Instituto Politécnico Nacional, Centro de Investigación en Ciencia Aplicada y Tecnología Avanzada, Cerro Blanco 141, Col. Colinas del Cimatario, Querétaro, Qro., Mexico. C.P. 76090. Reynaldo Pless-Elling, e-mail: rpless@ipn.mx
4 Unidad de Biotecnología. Centro de Investigación Científica de Yucatán. Calle 43 No. 130, Col. Chuburná de Hidalgo, Mérida, Yucatán, México, CP 97200.
Address Correspondence to: Aileen O'Connor-Sánchez, e-mail: aileen@cicy.mx
Recibido - Received 23.II.2011.
Aceptado - Accepted 15.V.2011.
Abstract. Mexico counts among the five countries with the highest biodiversity in the world. In the Yucatán Peninsula, there are aquatic ecosystems with a very special microbial diversity. These ecosystems are essential for the ecological equilibrium of the region, and are seriously threatened by human activities. Access and knowledge of the microbial resources of these environments have an enormous scientific interest, and could potentially result in biotechnological products which could lead to more efficient and environmentally friendly processes; it could also offer a full arsenal of microorganisms and/or novel molecules to the local and world industry to face the current needs. In recent years, with the rise of metagenomic technology, knowledge about microbial diversity in complex communities has started to develop astonishingly fast. This is because of the use of molecular methods which do not require culturing microorganisms in artificial media. Through DGGE, FISH, and TRFLP assays, 454 sequencing, Illumina sequencing, and functional analysis of metagenomic libraries, high-level scientific studies are being performed not only in microbiology, but also in other areas of knowledge. The results obtained thus far show that we are barely starting to perceive the enormous diversity present in the environments. The study and use of these resources claim for a high priority in the scientific policies of our country, and of other countries with which synergistic collaborations could be set up.
Keywords: Ecosystems; Biotechnology; 454-sequencing.
Resumen. México es uno de los cinco países con mayor biodiversidad en el mundo. En la Península de Yucatán existen ecosistemas acuáticos que contienen una diversidad microbiológica muy particular. Estos ambientes son fundamentales en el equilibrio ecológico de la zona, y están fuertemente amenazados debido a las actividades humanas. El acceso y conocimiento de los recursos microbianos de estos sitios, además de tener un enorme atractivo científico, tiene el potencial de traducirse en productos biotecnológicos que generen procesos más eficientes y ambientalmente amigables. Se puede poner a disposición de la industria no sólo local sino mundial, todo un arsenal de microorganismos y/o moléculas novedosas acordes con las necesidades actuales. En años recientes, con la aparición de la tecnología metagenómica, el conocimiento de la diversidad microbiana en comunidades complejas ha empezado a experimentar un desarrollo vertiginoso. Esto es debido al uso de métodos moleculares que no requieren el cultivo de microorganismos en medios artificiales. Mediante ensayos de DGGE, FISH, TRFLP, secuenciación 454, secuenciación Illumina y análisis funcionales de metagenotecas se están llevando a cabo estudios científicos del más alto nivel en microbiología, así como en otros campos del conocimiento. Los resultados obtenidos hasta ahora, muestran que apenas nos asomamos a la gigantesca y singular diversidad presente en los ambientes. El estudio y aprovechamiento de estos recursos debe ser un área prioritaria dentro de las políticas científicas de nuestro país y de los países con quienes podemos establecer colaboraciones sinérgicas.
Palabras clave: Ecosistemas; Biotecnología; Secuenciación 454.
INTRODUCTION
Mexico is one of the five countries with the highest biodiversity in the world; on its territory are from 8% to 12% of all the species in our planet. The aquifer, coastal lagoons and marshes, as well as the sea of the Yucatán peninsula, are key environments for the ecosystem of the south-east region of the country, due to their role in the ecology and their relation with the quality and level of the fresh water. The species living in these environments are part of an important trophic chain that begins with unicellular organisms and ends with man. Nevertheless, these systems are subject to an increasing anthropogenic pollution, mainly due to the increasing human population; the Rivera Maya is among the fastest growing areas in Latin America with an annual growth rate of approximately 20-25% (Meacham, 2007). As a result, these systems suffer alterations in their biodiversity and on the structure of the food chain that could produce important and synergistic changes in their equilibrium.
Water quality is being affected by physical, chemical, and biological factors (Metcalfe et al., 2010). Bacteria are among the most commonly occurring biological contaminants affecting the water bodies, and they can serve as indicators to assess the state of the ecosystem. For example, the fecal coliforms Escherichia coli and Enterococcus faecalis are correlated with fecal matter from humans and warm-blood animals, and their presence in the water may indicate the presence of enteropathogenic microorganisms. To date, there is no information on the (1) interactions between bacterial communities and microalgae in the aquifer of the Yucatán peninsula, (2) potential impact that the growth dynamics of these microorganisms might have on the trophic chain in this fragile ecosystem, and (3) water quality in different areas and environments.
Having access to the microbial resources in these unique environments may result in the formulation of biotechnological solutions that could result in cleaner and environmentally friendly processes, and may provide the local and worldwide industrial sphere with an arsenal of microorganisms and/or molecules resistant to the extreme conditions characteristic of these sites, such as pH, salinity, and redox potential.
For a long time, evaluation of microdiversity was limited to the study of microorganisms that could be cultured in the laboratory. However, in recent years, knowledge about the microbial diversity in complex communities had a marked development, due to the use of molecular methods which dispense with the need to culture the microorganisms in artificial, defined media. New molecular techniques have changed our view on microdiversity by enabling us to describe, monitor, and even control microbial communities. This allows us to obtain reliable data on the taxonomic identity of the microorganisms that colonize specific niches (Muyzer & Smalla, 1998; Sunnucks et al., 2000), to understand their spatial distribution (Cocolin et al., 2004), and to obtain quantitative data on the changes in the populations that result from accidental or external factors.
Microbial diversity as a source of wealth
For millennia, man has used microorganisms for his benefit in the preparation and/or conservation of products for human consumption. At the beginning, basic knowledge about many fermentative processes for the conservation and transformation of foodstuffs was acquired empirically and, certainly, in many instances by chance. As microbiological methods and artificial growth media developed, the use of microorganisms became simpler, and it became possible to standardize and understand many of the processes in which they were used. This spread the preparation of fermented products and other uses of microorganisms to areas beyond the food industry; they were used in the medical-pharmaceutical field, in the production of fine chemicals, in the treatment of residues, in biomining, in bioremediation, in agriculture, in biofuels, etc.
In 1990, Torsvik et al. published a pioneering work in which they showed, by DNA reassociation tests of singlestranded DNA, that the major part of the bacterial DNA in soil (in a deciduous forest in Norway) was highly heterogeneous, with a C0t1/2 value of 4600, which corresponds roughly to 4000 completely different bacterial genomes. Their results showed that the diversity of the bacterial population in the sample was so large that diversity indices could scarcely be determined by methods based on the heterogeneity of DNA. They concluded that the major fraction of the bacterial diversity resides in that part of the bacterial community which cannot be isolated and cultured with the standard techniques.
Nowadays, it is known that less than 1% of all of the microorganisms contained in an environmental sample can be found with the available culture methods; this situation is especially pronounced in complex ecosystems, where one gram may hold up to 109 prokaryotic cells, with an average of 2000 different genomes (Torsvik et al., 1990). The most plausible strategy to benefit from this wealth is to study specific ecosystems which could harbor microbial species or consortia with interesting activities, especially those which might form the basis for the development of clean bioprocesses (Langer et al., 2006). In order to advocate the conservation of such ecosystems it is necessary to present data which argue their importance; therefore it becomes imperative to determine their microbial diversity.
Many attempts have been made to improve microbial recovery in culture, mainly by a careful handling of the media and by increasing the number of species in the same culture (Kaeberlein et al., 2002); however, this has achieved only moderate success and the problem still remains an important challenge to microbiology. Kaeberlein and his coworkers (2002) attempted to grow non-cultivable organisms by supplying them with the chemical components of their natural habitat; they placed the microorganisms in diffusion chambers incubated under conditions resembling the native environment of these organisms to provide access to these components. In this manner they were able to recover about 40% of the original inoculums. This represents an important achievement when compared to attempts using established culture methodologies. In addition to this extraordinary "primary" recovery in culture, they attempted to isolate colonies from the cultivable community. However, the isolates did not grow in most cases (86%), and those which did grow proved to be mixed cultures. Only a few, fast growing colonies were pure, and they represented only 0.054% of the original inoculums. These data lead to interesting conclusions on the ecology of microorganisms, emphasizing the possible importance of specific signals coming from neighboring organisms, which might convey the notion of a familiar environment. This would underline the importance of the total community or consortium for the success of any individual member of the community. This implies that the microorganisms will not grow in an unfamiliar environment (lacking their usual neighbors), even in the presence of the appropriate nutrients. It may be possible that such nutrients may not be directly available for certain microorganisms, and may require some type of prior transformation (oxidation/ reduction, unfolding) by another group of microorganisms. For these reasons, lack of knowledge about the current microorganisms, and the poor understanding of the requirements for their successful cultivation may combine to constitute a vicious circle, frustrating any progress in this area (Fig. 1).
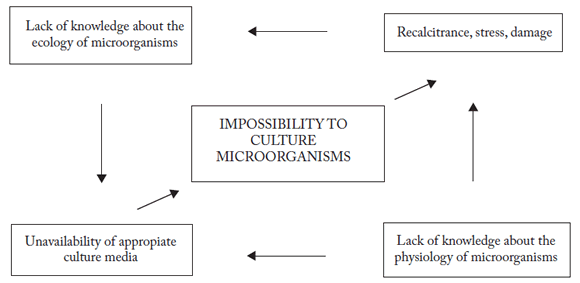
Fig. 1. The vicious circle of the impossibility to cultivate most microorganisms.
Fig. 1. El círculo vicioso de la imposibilidad de cultivar la mayoría de los microorganismos.
The aquifer of Yucatán peninsula
This peninsula contains the most extensive system of underground rivers in the world. The great biological diversity of the region is based on this reserve of hidden water, whose special characteristics and importance can only be understood based on its geological history. Geologically speaking, the Yucatán peninsula is one of the youngest regions of Mexico; its present shape dates back only ten-thousand years. During the Mesozoic era it was part of the ocean floor; as a result, it was made up largely from limestone and evaporite rocks. During the glacial and interglacial periods of the Pleistocene it arose from the sea. During this process, the chemical dissolution of the limestone at the boundary between the fresh (from rain) and salty waters (from the sea) led to the formation of an extensive system of caverns, which constitute a highly permeable medium (Rebolledo-Vieyra, 2005). As underwater limestone continually dissolves in the passing flow, and as calcium carbonate in air-free conditions precipitates, an additional feature is that the shape of the caverns behaves as a dynamic system.
Sixty-five million years ago, the impact of the meteorite that caused one of the most massive extinctions of species in the history of life, marked the Yucatán peninsula with a very extensive system of semi-circular fault lines. This gave rise to the formation of one of the most uncommon hydrological systems in the world, the so-called "cenotes-ring of Chicxulub". Cenotes are originally underground caverns filled with water, which then opened up to the earth´s surface when their roof partially collapsed. The water in these cenotes can be at different levels, and, as in the rest of the aquifer, it usually has two layers: an upper layer of fresh water fed by rain, and a deeper layer of salt water, fed from the sea, which is termed "saline intrusion".
There is evidence for the existence of cenotes and portions of the aquifer that can still be considered pristine, in the sense of not having had any significant contact with human beings, or with their waste or other products. There are very few studies of the flora and fauna of the aquifer, and virtually nothing has been published on its microflora. Nonetheless, there have been reports on endemic species, mainly on fish and arthropods.
The unique composition and conformation of the subsoil, the rich biological diversity of the region in general, the lack of study of this biodiversity (of the aquifer in particular), and the highly isolated nature of some of its areas, make this an environment of great interest for metagenomic analyses.
The microorganisms and the sea
The fresh water of the aquifer flows toward the sea and emerges, either on the coast or offshore, in locations where it creates areas of unique physical, chemical, and biological characteristics. The lagoons which are formed and the areas of offshore discharge also contain a highly interesting microflora.
It is known that the evolutionary history of the microorganisms goes back more than three-and-a-half billion years, and that it has taken place to an important extent in a marine medium. Since then, the microorganisms are an essential part of the trophic networks in oceanic ecosystems. A wealth of information about them has been uncovered in recent years by metagenomics, such as an incredible diversity, vast swathes of uncharacterized metabolism, increased complexity of biogeochemical pathways, and even some paradigm shifts in our understanding of marine microbial ecology (Gilbert & Dupont, 2011).
The oceanic environment covers 70% of the planet's surface, and the average depth of the oceans is 4000 m; it is therefore obvious that the study of prokaryote communities resident in the oceans is of outstanding importance for our knowledge of (1) biodiversity on a global scale, and (2) the biogeochemical cycles. One can estimate that the oceans contain more prokaryotic cells than any other aqueous environment, and that they are, by biomass, the most productive ecosystems in our planet; nonetheless, to date, they have not been subjected to the study they deserve as an extraordinary source of new compounds.
In 2007, Rusch et al. reported a metagenomic study of the marine planktonic microbiota in which surface (mostly marine) water samples were analyzed as part of the so called "Sorcerer II Global Ocean Sampling (GOS) expedition". Their samples, collected across a several-thousand km transect from the North Atlantic through the Panama Canal, and ending in the South Pacific, yielded an extensive dataset. It consisted of 7.7 million sequencing reads (6.3 billion bp). They found that although a few major microbial clades dominated the planktonic marine niche, the dataset contained great diversity, with 85% of the assembled sequence and 57% of the unassembled data being unique at a 98% sequence identity cutoff. Using the metadata associated with each sample and sequencing library, they developed new comparative genomic and assembly methods. One comparative genomic method, termed ''fragment recruitment'', addressed questions of genome structure, evolution, and taxonomic or phylogenetic diversity, as well as the biochemical diversity of genes and gene families. A second method, termed ''extreme assembly,'' made possible the assembly and reconstruction of large segments of abundant but clearly nonclonal organisms. Within all abundant populations analyzed, they found extensive intra-ribotype diversity in several forms: (1) extensive sequence variation within orthologous regions throughout a given genome; despite coverage of individual ribotypes approaching 500-fold, most individual sequencing reads were unique; (2) numerous changes in gene content, some with direct adaptive implications; and (3) hypervariable genomic islands that are too variable to assemble. The intra-ribotype diversity is organized into genetically isolated populations that have overlapping but independent distributions, implying distinct environmental preferences. They presented novel methods for measuring the genomic similarity between metagenomic samples, and showed how they may be grouped into several community types. Specific functional adaptations were identified both within individual ribotypes and across the entire community, including proteorhodopsin spectral tuning and the presence or absence of the phosphatebinding gene PstS.
In the same year, Yooseph et al. (2007) reported the use of sequence similarity clustering to explore proteins with a comprehensive dataset consisting of sequences from databases, together with 6.12 million proteins predicted from an assembly of 7.7 million GOS sequences. They found that the GOS dataset covered nearly all known prokaryotic protein families. A total of 3995 medium- and large-sized clusters consisting of only GOS sequences were identified, out of which 1,700 had no detectable homology to known families. The GOS-only clusters contained a higher than expected proportion of sequences of viral origin, thus reflecting a poor sampling of viral diversity to date. Protein domain distributions in the GOS dataset, and protein databases showed distinct biases. Several protein domains that were previously categorized as kingdom specific were shown to have GOS examples in other kingdoms. About 6000 sequences (ORFans) from the literature lacked similarity to known proteins. Their analysis indicated that new families were discovered at a rate that was linear or almost linear to the addition of new sequences, implying that we are still far from discovering all protein families in nature.
From all this, it is presumable that the oceans are a reservoir of molecules and compounds with potential use in basic research, in industry, in biomedicine, in bioremediation, etc. This is because they are environments of great chemical diversity which has hardly been studied, with unique conditions quite distinct from land environments.
In this environment, several interesting examples of food chains have been found, in which bacteria play important roles both at the beginning and end (Duffy, 2002). This is because they contribute to the production of foodstuffs starting from organic substrates dissolved in the water, and also carry out the mineralization of the organic matter, thereby returning the nutrients to the medium (Azam & Worden, 2004). On the seafloor, they also perform tasks which we are only starting to understand, forming part of chains which entail oxidation/reduction of inorganic molecules. These discoveries have been surprising, as the sea is an environment which has barely been studied by microbiologists because it was thought to hold little bacterial diversity at its coasts, and hardly any bacteria in deeper regions offshore, as a result of high salinity, weak or absent light and drastic shortage of oxygen and nutrients. Nevertheless, a great biodiversity has been found at the coasts, because oxygen is highly available near the surface, and the proximity of the land assures a wealth of nutrients coming from the organic matter in the coastal vegetation. In these ecosystems, photosynthetic and aerobic microorganisms predominate, e.g. microalgae and cyanobacteria.
Other prokaryotes found in oceanic environments belong to the domain Archaea, which were known to reside exclusively in very special environments; for instance, some Archaea, thought to be found only in extremely hot environments (Cenarchaeum symbiosum) have closely related species inhabiting Antarctic waters, at -1.5 °C (DeLong, 1997). In the absence of currents, on the seafloor close to the coast, water is less rich in oxygen; consequently, in this environment mainly anaerobic photosynthetic bacteria can be found. This is particularly marked in coastal wetlands such as those found along the coast of the Yucatán peninsula.
Culture-independent microbiology
Metagenomics can be defined as the analysis, usually by sequencing and/or cloning, of DNA extracted directly from the environment, without passing through a stage of culturing
of the organisms contained in a sample. It is also sometimes referred to as "environmental genomics".
The use of the methodologies of Genomics has led to a better and deeper knowledge of both basic microbial ecology (i.e., the relations between the microorganisms and the environment) and applied microbial ecology (public health, agriculture, aquaculture, industry, etc). As a result, the study of microbial ecology has expanded very rapidly in recent years, mainly describing microbial diversity and characterizing microbial activities ( Luna et al., 2002; Bouillon et al., 2004). These studies have provided important information in various areas of microbiology and biotechnology, and have set the bases for (1) the control of undesirable microorganisms, and (2) the use of microorganisms and their products for human benefits, in bioprocesses which provide goods and services (Melcher et al., 2002; Lors et al., 2004) .
The description of the microorganisms present in environmental samples has seen great advances thanks to the use of the gene sequences coding for ribosomal RNA, above all of the gene coding for the minor subunit of the bacterial ribosome. This molecule is present in all organisms, and performs the same function in all of them. The method can be used to perform comparisons at different levels of resolution, even though different regions of the 16S rRNA molecule have different sequence variability between different taxa. Sequence analysis of these genes makes it possible to define the phylogenetic relationships between the microorganisms. This has led to a restructuring of modern taxonomy (Woese et al., 1990; Woese, 1998).
DGGE and TGGE
The use of molecular tools such as DGGE (denaturinggradient gel electrophoresis), TGGE (temperature-gradient gel electrophoresis), and FISH (fluorescent in situ hybridization) has greatly expanded in taxonomic studies. This allowed to gain greater knowledge of bacterial diversity and of the structure of bacterial communities in different environments., During the last decade, these techniques have also allowed the use of functional genes as molecular markers to relate them with the structure and function of microbial communities (Muyzer & Smalla, 1998).
The DGGE technique generates precise taxonomic and phylogenetic information, which allows the study of the structure of bacterial communities and biofilms based on the denaturalization of short (200-700bp) fragments of genomic DNA collected from the environment (Muyzer & Smalla, 1998). The DNA is extracted from environmental samples, and segments of the 16S rRNA gene are amplified by PCR. The resulting products are all of the same size, but they have different sequences; this is because they come from all the different bacteria which were present in the sample. The doublestranded DNA amplicons are then resolved by electrophoresis through a polyacrylamide gel containing a denaturing gradient, which produces a pattern that reflects the genetic diversity of the bacterial community in the original sample (LaPara et al., 2002). Furthermore, it is possible to identify taxa on the basis of this electrophoresis, by recovering each band from the gel and sequencing it, for a correlation with known sequences in data bases.
TRFLP
Methods to generate genetic fingerprints of entire communities, such as the analysis of terminal transcription fragment length polymorphisms [TRFLP, Liu et al. (1997)] have been used frequently to compare microbial communities. This has been because the techniques involved are relatively rapid, of low cost, and can be used in a massively parallel manner. TRFLP is a technique used to study complex communities based on variations in the 16S rRNA genes (Osborn et al., 2000). It can be used to study either the structure of communities, or their dynamic response to changes in their environment. The technique has been applied to the study of microbial communities in various environments, such as the soil (Derakshani et al., 2001) and wastewater systems (Eschenhagen et al., 2003), and the characterization of the bacterial mouth flora of healthy persons and of patients suffering from periodontitis (Sakamoto et al., 2003).
TRFLP analysis has the advantages of being a cultureindependent method, relatively fast, of low cost, and useful for sampling the diversity of complex communities without requiring knowledge of the genomic sequences involved. The disadvantages of this method are mainly that it may produce different results when applied to communities with very high diversity (Orcutt et al., 2009), and that it may fail to detect taxa at low or rare organism abundance (Engebretson& Moyer, 2003; Bent et al., 2007).
Pyrosequencing
Another molecular technique that is being used ever more frequently in microbial diversity studies is pyrosequencing, also known as 454 sequencing. This technology provides an ultra-massive DNA sequencing system based on chemiluminescence. With this system, genomes and metagenomes can be sequenced or resequenced, and tagging of DNA sequences of special interest is also possible, all in record time. The present model of the instrument, named "Genome Sequencer FLX - GS FLX Titanium" generates 400 million bases of sequence in only 10 hours. This technology has set off hundreds of scientific studies of the highest level, not only in microbiology, but also in various other fields of knowledge, such as cancer research, infectious diseases, drug discovery anthropology, and paleontology.
To give an idea of how recently this sequencing technology was introduced, we need only state that "454 Life Sciences" was founded in the year 2000. October 2005 saw the release to market of the "Genome Sequencer 20", which was the first system of the next generation. In November 2006, the first million base pairs of the sequence of Homo neanderthalensis, obtained by this method, were published in Nature, in collaboration with Svante Pääbo. The next model, "Genome Sequencer FLX Titanium", released in October 2008, can produce one million 400-base-pair reads in one run (www.454.com). One of the main goals of metagenomic studies is to gain an unbiased picture of the phylogenetic composition and the functional diversity within a given microbial community. Even though technical and economic limitations still preclude the full depth which may be desirable in some analyses, especially in the study of metabolic profiles and adaptive dynamics, at present the Genome Sequencer FLX stands for the most advanced technology available to overcome these barriers.
Metagenomic samples can be taken from virtually any site; this includes microenvironments of the human body, extreme environments, highly contaminated environments, different locations in the water column of the ocean, etc. Some of the possible applications of the 454 technology include: (i) calculate the relative abundance of microbial species under different environmental conditions; one can count the gene tags (sequences that are specific for one species or one gene), dispensing cloning steps; (ii) determine the genetic content, detect genes more quickly and make predictions as to function, as long and precise reads are possible; (iii) analyze the regulation and expression dynamics of genes in various environments, metatranscriptomic expression profiling and functional annotation can be performed; (iv) analyze viral infections quickly and precisely; one can sequence RNA fragments amplified from material obtained from infected individuals, and (v) establish the "signature" or profile that characterizes a microbial community; one can identify its members and predict their functions and relative abundance, all in one run.
The recent publications on metagenomes and bacterial genomes have started to show the immense diversity of the microbial world. For instance, a sampling of marine viral genomes at 68 sites indicated that the global diversity of viruses could be of the order of hundreds of thousands of species (Angly et al., 2006). In another sampling, it was found that two deep-mine samples held widely different communities, with different metabolisms, even when the samples had been collected at very closely neighboring sites. In addition, it is interesting to note that the metagenomes of the two sites were completely different from those of other communities which had been sequenced earlier (Edwards et al., 2006). A third publication, dealing with marine microbial diversity, described an approach using ribosomal RNA sequence tags and estimated a diversity three orders of magnitude greater than the earlier calculation of 106 species (Sogin et al., 2006).
While in the majority of the cases the role played by most of the microorganisms has not been elucidated, several recent publications have shown that they maintain mutual interactions of competence and synergy, and that these interactions can be altered in response to changes in the environment. For instance, a study of the human digestive tract showed the presence of two main populations of bacteria, the Bacteroidetes and the Firmicutes, whose relative abundance fluctuates as the body fat of the individual changes (Turnbaugh et al., 2009).
Some examples of recent metagenomic work performed with the 454 technology are (1) the exploration of the deepest phreatic trough in the world (Sahl et al., 2010), and (2) studies on the structure of microbial communities in soil and hypersaline sediment gradients (Hollister et al., 2010) on bacterial communities of the Arctic Ocean (Kirchman et al., 2010) and on the seasonal dynamics of bacterio-plankton in the Baltic Sea (Andersson et al., 2010).
Access to microbial resources
The advances made in molecular biology and genomics, and their application to prokaryote biology have set the bases not only for sequencing, but also for cloning and the functional analysis of metagenomes (Rondon et al., 1999). This allows not only to study those microorganisms which could heretofore not be cloned, but also to clone segments of their DNA sufficiently long as to contain complete operons, which are able to direct the synthesis of complex molecules (Rondon et al., 2000; Schloss & Handelsman, 2003); in prokaryotes the genes which relate to a given metabolic pathway are usually grouped in clusters. Also, in cases where the end product of a given route may be potentially toxic to the host cell, this methodology enables the simultaneous cloning (on one and the same DNA segment) of the resistance genes, which are usually located close to the genes for the metabolic pathway.
Ever since streptomycin was discovered as a bioproduct of actynomycetes, in the first half of last century, microorganisms have been frequently studied in the soil, in search of active principles. These studies always suffered from difficulties in culturing the microorganisms, which has led to numerous repeated reisolations (Handelsman et al., 1998). Nowadays, soils, bodies of water, and extreme environments are seen as a rich and largely unexplored source in the search for new products (Daniel, 2004); therefore, the methodologies which aim at their metagenomic exploration are important strategies for accessing the functional diversity of their as yet uncultured microorganisms (Courtois et al., 2003).
Recent years have seen a growing activity in the metagenomics of marine microorganisms (Martín-Cuadrado et al., 2007) and of extreme environments (extremophiles), as well as on the metabolic adjustments which they must have undergone to survive in such environments. Rothschild& Mancinelli (2001) have made them the subject of numerous studies, because of the potential usefulness of the organisms themselves or of their enzymes in biotechnological or industrial processes. A classical example is the isolation of Thermus aquaticus, a hyperthermophilic organism which lives in hot springs at temperatures above 70 °C. Its thermostable DNA polymerase (Taq) has revolutionized biotechnology, medicine, and other life sciences, as it is a main actor in the amplification of DNA by PCR (polymerase chain reaction) (Saiki et al., 1985, 1988).
Constructing metagenomic gene libraries requires DNA fragments of relatively large size (30-50 kb), as this improves the likelihood to clone the genes necessary for the synthesis of a given product. This is especially important in the case of complex samples such as those collected from soil, sediments or other environments containing a large concentration of decomposing organic matter, as many of the compounds present in such samples may copurify with the DNA (e.g., humic acids or fulvic acids) and may be inhibitory to the various enzymes which are used in cloning (Wilson, 1997).
Two methods of analysis have been proposed to screen metagenomic libraries: (i) functional analysis, which relies on the identification of clones that express specific traits (enzymatic activities, antimicrobial activity, presence of certain metabolites, etc.), and (ii) analysis based on nucleotide sequence, which uses conserved sequences to design hybridization probes. Both approaches have been used successfully (Handelsman, 2004; Grant & Heaphy, 2010; Kapardar et al., 2010; Lee et al., 2010).
It is also possible to screen the genomic library searching for sequences which contain philogenetic information (such as the 16S rRNA gene) and which afford data on the origin of the heterologous DNA. This allows to establish the relation between the phylogeny of the as yet uncultured microorganisms and their genetic and physiologic activities coded in their inserts (Rondon et al., 2000; Beja et al., 2000; Béjà, 2004).
Although metagenomics is a very young science, it has to date provided results that justify the intense effort which is now being expended in this field. For instance, the year 2002 saw the isolation of turbominin A and turbominin B starting from a metagenomic soil library built in a BAC vector. These two compounds proved to be wide-spectrum antibiotics, active against gram-positive and gram-negative bacteria (Rondon et al., 2000; Gillespie et al., 2002). This, together with the isolation of the operon for biotin biosynthesis (Entcheva et al., 2001), showed the usefulness of metagenomic studies in the search for new natural products of benefit for mankind. It was also demonstrated that it is possible to clone entire metabolic pathways starting from a metagenome and its expression in heterologous systems. Other authors have reported the isolation of enzymes with different activities of biotechnological interest: chitinases, identified in metagenomic libraries of coastal and estuarine waters (Cottrell et al., 1999); 4-hydroxybutyrate dehydrogenase, isolated from a metagenomic E. coli clone able to grow in a medium containing 4-hydroxybutyrate as the only carbon source (Henne et al., 1999); clones with lipolytic activity isolated from a soil metabolic library, one of which genes turned out to be a lipase and other two showed homology with esterases (Henne et al., 2000); polyketide synthase (an enzyme involved in the synthesis of a compound with antitumor activity) from a soil metagenome and from microorganisms associated with the sea sponge Discodermia dissoluta (Schirmer et al., 2005); an amylase with a pH optimum of 9.0 from the soil metagenome (Yun et al., 2004), and a thermostable esterase isolated from a metagenomic library obtained from the DNA in sediments and muds from a hot spring, which proved to be active from 30 °C to 95 °C (Rhee et al., 2005).
Obtaining hydrolytic enzymes of microbial origin is very important for industrial applications, especially in the case of those enzymes which may be a useful adjunct in the transformation of agricultural residues for purposes of energy production and/or synthesis of high value-added chemicals. For instance, the treatment of this type of residue in bioconversion processes may require the degradation of the lignocellulosic materials contained in the plant material. Degradation of lignin is an oxidative process involving enzymes such as lignin peroxidases (LiP), manganese peroxidases (MnP), and laccases. These enzymes have been widely studied in different microorganisms; however, their hydrolytic activities are subject to a number of physical and chemical factors that limit their application in the industrial sphere. There is thus a need to find new enzymes with increased stability for (1) extreme pH values, (2) the presence of organic solvents or of ionic detergents, or (3) elevated salt content or temperatures. Proteolytic and amylolytic enzymes are also widely used in the food industry, as well as in bioconversion and refineries.
Outlook and potential for microbial diversity
Recent years have seen a growing interest in marine microorganisms and extremophiles, and the adjustments that these species had to undergo in their metabolism to survive in these environments (Rothschild & Mancinelli, 2001). This makes them interesting subjects for studying because of their potential application in biotechnological and industrial processes. The results obtained to date from metagenomic studies show that we have barely started to understand the enormous metabolic diversity which is present in the environment (Béjà, 2004). This diversity can be a source of new products, genes and enzymes of biotechnological importance (Courtois et al., 2003; Venter et al., 2004; DeLong& Karl, 2005; Foti et al., 2007). Nevertheless, many of these enzymes, while detectable in environmental samples, could not be used as attempts to culture the species producing them were unsuccessful. The use of microbial consortia has emerged as a possible biotechnological strategy (Manonmani et al., 2000, Schlotelburg et al., 2002), but microbiology still needs to solve the problem of how to culture such consortia in a way that they maintain the necessary structure and dynamics. Knowledge of the taxonomic structure and population dynamics of these consortia, obtained by metagenomic techniques, sets the basis for understanding the mechanisms which control their function, and for designing strategies for their exploitation.
Given the richness of ecosystems in Mexico, the microbial diversity resident in these niches may hold the solution for many of the problems that we face today, both on a national and on a global level. The study and exploitation of these resources should be a primary aim of the scientific policies of our country (Mexico), as well as of other countries with which synergic collaborations would be advantageous.
REFERENCES
1. Andersson, A.F., L. Riemann & S. Bertilsson (2010). Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J 4: 171-181. [ Links ]
2. Angly, F.E., B. Felts, M. Breitbart, P. Salamon, R.A. Edwards, C. Carlson, A.M. Chan, M. Haynes, S. Kelley, H. Liu, J.M. Mahaffy, J.E. Mueller, J. Nulton, R. Olson, R. Parsons, S. Rayhawk, C.A. Suttle & F. Rohwer (2006). The Marine Viromes of Four Oceanic Regions. PLoS Biology 4: e368. doi:10.1371/journal. pbio.0040368. [ Links ]
3. Azam, F. & A.Z. Worden (2004). Microbes, Molecules, and Marine Ecosystems. Science 303: 1622-1624. [ Links ]
4. Béjà, O. (2004). To BAC or not to BAC: marine ecogenomics. Current Opinion in Biotechnology 15: 187-190. [ Links ]
5. Béjà, O., L. Aravind, E.V. Koonin, M.T. Suzuki, A. Hadd, L.P. Nguyen, S.B. Jovanovich, C.M. Gates, R.A. Feldman, J.L. Spudich, E.N. Spudich & E.F. DeLong (2000). Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science 289: 1902-1906. [ Links ]
6. Bent, S.J., J.D. Pierson & L.J. Forney (2007). Measuring species richness based on microbial community fingerprints: The emperor has no clothes. Appl Environ Microbiol 73: 2399-2401. [ Links ]
7. Bouillon, S., T. Moens, N. Koedam, F. Dahdouh-Guebas, W. Baeyens & F. Dehairs (2004). Variability in the origin of carbon substrates for bacterial communities in mangrove sediments. FEMS Microbiology Ecology 49: 171-179. [ Links ]
8. Cocolin, L., K. Rantsiou, L. Iacumin, R. Urso, C. Cantoni & G. Comi (2004). Study of the ecology of fresh sausages and characterization of populations of lactic acid bacteria by molecular methods. Appl Environ Microbiol 70: 1883-1894. [ Links ]
9. Cottrell, M.T., J.A. Moore & D.L. Kirchman (1999). Chitinases from Uncultured Marine Microorganisms. Applied and Environmental Microbiology 65: 2553-2557. [ Links ]
10. Courtois, S., C.M. Cappellano, M. Ball, F.X. Francou, P. Normand, G. Helynck, A. Martínez, S.J. Kolvek, J. Hopke, M.S. Osburne, P.R. August, R. Nalin, M. Guérineau, P. Jeannin, P. Simonet & J.L. Pernodet (2003). Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Applied Environmental Microbiology 69: 49-55. [ Links ]
11. Daniel, R. (2004). The soil metagenome - a rich resource for the discovery of novel natural products. Current Opinion in Biotechnology 15: 199-204. [ Links ]
12. DeLong, E. (1997). Marine microbial diversity: The tip of the iceberg. Trends in Biotechnology 15: 203-207. [ Links ]
13. DeLong, E. & D. Karl (2005). Genomic perspectives in microbial oceanography. Nature 437: 336-342. [ Links ]
14. Derakshani, M., T. Lukow & W. Liesack (2001). Novel Bacterial Lineages at the (Sub)Division Level as Detected by Signature Nucleotide-Targeted Recovery of 16S rRNA Genes from Bulk Soil and Rice Roots of Flooded Rice Microcosms. Applied and Environmental Microbiology 67: 623-631. [ Links ]
15. Duffy, J.E. (2002). Biodiversity and ecosystem function: the consumer connection. Oikos 99: 201-219. [ Links ]
16. Edwards, R.A., B. Rodriguez-Brito, L. Wegley, M. Haynes, M. Breitbart, D.M. Peterson, M.O. Saar, S. Alexander, E.C. Alexander & F. Rohwer (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7: 57. [ Links ]
17. Engebretson, J.J. & C.L. Moyer (2003). Fidelity of select restriction endonucleases in determining microbial diversity by terminal-restriction fragment length polymorphism. Applied Environmental Microbiology 69: 4823-4829. [ Links ]
18. Entcheva, P., W. Liebl, A. Johann, T. Hartsch & W. Streit (2001). Direct cloning from enrichment cultures, a reliable strategy for isolation of complete operons and genes from microbial consortia. Applied Environmental Microbiology 67: 89-99. [ Links ]
19. Eschenhagen, M., M. Schuppler & I. Röske (2003). Molecular characterization of the microbial community structure in two activated sludge systems for the advanced treatment of domestic effluents. Water Research 37: 3224-3232. [ Links ]
20. Foti, M., D. Sorokin, B. Lomans, M. Mussman, E. Zacharova, N.V. Pimenov, J. Kuenen & G. Muyzer (2007). Diversity, Activity, and Abundance of Sulfate-Reducing Bacteria in Saline and Hypersaline Soda Lakes. Applied Environmental Microbiology 73: 2093- 2100. [ Links ]
21. Gilbert, J.A. & C.L. Dupont (2011). Microbial Metagenomics: Beyond the Genome. Annual Review of Marine Science 01: 347-371. [ Links ]
22. Gillespie, D.E., S.F. Brady, A.D. Bettermann, N.P. Cianciotto, M.R. Liles, M.R. Rondon, J. Clardy, R.M. Goodman & J. Handelsman (2002). Isolation of antibiotics turbomycin a and b from a metagenomic library of soil microbial DNA. Applied Environmental Microbiology 68: 4301-4306. [ Links ]
23. Grant, W.D. & S. Heaphy (2010). Metagenomics and recovery of enzyme genes from alkaline saline environments. Environmental Technology 31: 1135-1143. [ Links ]
24. Handelsman, J., M. Rondon, S. Brady, J. Clardy & R. Goodman (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chemistry and Biology 5: R245-R249. [ Links ]
25. Handelsman, J. (2004). Metagenomics: Application of Genomics to Uncultured Microorganisms. Microbiology and Molecular Biology Review 68: 669-685. [ Links ]
26. Henne, A., R. Daniel, R. Schmitz & G. Gottschalk (1999). Construction of environmental DNA libraries in Escherichia coli and screening for the presence of genes conferring utilization of 4-hydroxybutyrate. Applied Environmental Microbiology 65: 3901-3907. [ Links ]
27. Henne, A., R. Schmitz, M. Bomeke, G. Gottschalk & R. Daniel (2000). Screening of environmental DNA libraries for the presence of genes conferring lipolytic activity on Escherichia coli. Applied Environmental Microbiology 66: 3113-3116. [ Links ]
28. Hollister, E.B., A.S. Engledow, A.J. Hammett, T.L. Provin, H.H. Wilkinson & T.J. Gentry (2010). Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4: 829-838. [ Links ]
29. Kaeberlein, T., K. Lewis & S.S Epstein (2002). Isolating "Uncultivable" Microorganisms in Pure Culture in a Simulated Natural Environment. Science 296: 1127-1129. [ Links ]
30. Kapardar, R.K., R. Ranjan, A. Grover, M. Puri & R. Sharma (2010). Identification and characterization of genes conferring salt tolerance to Escherichia coli from pond water metagenome. Bioresource Technology 101: 3917-3924. [ Links ]
31. Kirchman, D.L., M.T. Cottrell & C. Lovejoy (2010). The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environmental Microbiology 12: 1132-1143. [ Links ]
32. Langer, M., E. Gabor, K. Liebeton, G. Meurer, F. Niehaus, R. Schulze, J. Eck & P. Lorenz (2006). Metagenomics: An inexhaustible access to nature's diversity. Biotechnology Journal 1: 815-821. [ Links ]
33. Liu, W., T. Marsh, H. Cheng & L. Forney (1997). Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Applied Environmental Microbiology 63: 4516-4522. [ Links ]
34. LaPara, T.M., C.H. Nakatsu, L.M. Pantea & J.E. Alleman (2002). Stability of the bacterial communities supported by a seven-stage biological process treating pharmaceutical wastewater as revealed by PCR-DGGE. Water Research 36: 638-646. [ Links ]
35. Lee, H.S., K.K. Kwon, S.G. Kang, S.S. Cha, S.J. Kim & J.H. Lee (2010). Approaches for novel enzyme discovery from marine environments. Current Opinion in Biotechnology 21: 353-357. [ Links ]
36. Lors, C., C. Tiffreau & A. Laboudigue (2004). Effects of bacterial activities on the release of heavy metals from contaminated dredged sediments. Chemosphere 56: 619-630. [ Links ]
37. Luna, G., E. Manini & R. Danovaro (2002). Large fraction of dead and inactive bacteria in coastal marine sediments: comparison of protocols for determination and ecological significance. Applied Environmental Microbiology 68: 3509-3513. [ Links ]
38. Manonmani, H., D. Chandrashekaraiah, N. Reddy, C. Elcey & A. Kunhi (2000). Isolation and Acclimation of a Microbial Consortium for Improved Aerobic Degradation of a-Hexachlorocyclohexane. Journal of Agricultural and Food Chemistry 48: 4341-4351. [ Links ]
39. Martín-Cuadrado, A.B., P. López-García, J.C. Alba, D. Moreira, L. Monticelli et al. (2007). Metagenomics of the Deep Mediterranean, a Warm Bathypelagic Habitat. PLoS ONE 2: e914. [ Links ]
40. Meacham, S. (2007) Freshwater Resources in the Yucatan Peninsula. Sustainable Management of Groundwater in Mexico: Proceedings of a Workshop (Series: Strengthening Science-Based Decision Making in Developing Countries) http://www.nap.edu/ catalog/11875.html [ Links ]
41. Melcher, R., S. Apitz & B. Hemmingsen (2002). Impact of Irradiation and Polycyclic Aromatic Hydrocarbon Spiking on Microbial Populations in Marine Sediment for Future Aging and Biodegradability Studies. Applied Environmental Microbiology 68: 2858-2868. [ Links ]
42. Metcalfe, C.D., P.A. Beddows, G. Gold-Bouchot, T.L. Metcalfe, H. Li & H. VanLavieren (2010). Contaminants in the coastal karst aquifer system along the Caribbean coast of the Yucatan Peninsula, Mexico Environmental Pollution 159:991-997. [ Links ]
43. Muyzer, G. & K. Smalla (1998). Application of denaturing gradient gel electrophoresis (DGGE) and temperature gradient gel electrophoresis (TGGE) in microbial ecology. Antonie van Leeuwenhoek 73: 127-141. [ Links ]
44. Orcutt, B., B. Bailey, H. Staudigel, B.M. Tebo & K.J. Edwards (2009). An interlaboratory comparison of 16S rRNA gene-based terminal restriction fragment length polymorphism and sequencing methods for assessing microbial diversity of seafloor basalts. Environmental Microbiology 11: 1728-1735. [ Links ]
45. Osborn, A.M., E.R.B. Moore & K.N. Timmis (2000). An evaluation of terminal-restriction fragment length polymorphism (TRFLP) analysis for the study of microbial community structure and dynamics. Environmental Microbiology 2: 39-50. [ Links ]
46. Rebolledo-Vieyra, M. (2005). Breve historia geológica de la península de Yucatán y el origen de los cenotes. En: Medina, A. (ed) "Agua, rocas y siglos", Ed. Pixel, México. [ Links ]
47. Rhee, J., D. Ahn, Y. Kim & J. Oh (2005). New thermophilic and thermostable esterase with sequence similarity to the hormonesensitive lipase family, cloned from a metagenomic library. Applied Environmental Microbiology 71: 817-825. [ Links ]
48. Rondon, M.R., P.R. August, A.D. Bettermann, S.F. Brady, T.H. Grossman, M.R. Liles, K.A. Loiacono, B.A. Lynch, I.A. Macneil, C. Minor, C.L. Tiong, M. Gilman, M.S. Osburne, J. Clardy, J. Handelsman & R.M. Goodman (2000). Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Applied Environmental Microbiology 66: 2541-2547. [ Links ]
49. Rondon, M.R., S.J. Raffel, R.M. Goodman & J. Handelsman (1999). Toward functional genomics in bacteria: Analysis of gene expression in Escherichia coli from a bacterial artificial chromosome library of Bacillus cereus. Proceedings of the National Academy of Sciences USA 96: 6451-6455. [ Links ]
50. Rothschild, L. & R. Mancinelli (2001). Life in extreme environments. Nature 409: 1092-1101. [ Links ]
51. Rusch, D.B., A.L. Halpern, G. Sutton, K.B. Heidelberg, S. Williamson, S. Yooseph, D. Wu, J.A. Eisen, J.M. Hoffman, K. Remington, K. Beeson, B. Tran, H. Smith, H. Baden-Tillson, C. Stewart, J. Thorpe, J. Freeman, C. Andrews-Pfannkoch, J.E. Venter, K. Li, S. Kravitz, J.F. Heidelberg, T. Utterback, Y.H. Rogers, L.I. Falcón, V. Souza, G. Bonilla-Rosso, L.E. Eguiarte, D.M. Karl, S. Sathyendranath, T. Platt, E. Bermingham, V. Gallardo, G. Tamayo-Castillo, M.R. Ferrari, R.L. Strausberg, K. Nealson, R. Friedman, M. Frazier & J.C. Venter (2007). The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biology 5: 398-431. [ Links ]
52. Sahl, J.W., N. Fairfield, J.K. Harris, D. Wettergreen, W.C. Stone & J.R. Spear (2010). Novel microbial diversity retrieved by autonomous robotic exploration of the world's deepest vertical phreatic sinkhole. Astrobiology 10: 201-213. [ Links ]
53. Saiki, R., S. Scharf, F. Faloona, K. Mullis, G. Horn, H. Erlich & N. Arnheim (1985). Enzymatic amplification of b-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230: 1350-1354. [ Links ]
54. Saiki, R., D. Gelfand, S. Stoffel, S. Scharf, R. Higuchi, G. Horn, K. Mullis & H. Erlich (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239: 487-491. [ Links ]
55. Sakamoto, M., Y. Takeuchi, M. Umeda, I. Ishikawa & Y. Benno (2003). Application of terminal RFLP analysis to characterize oral bacterial flora in saliva of healthy subjects and patients with periodontitis. Journal of Medical Microbiology 52: 79-89. [ Links ]
56. Schirmer, A., R. Gadkari, C. Reeves, F. Ibrahim, E. DeLong& C. Hutchinson (2005). Metagenomic analysis reveals diverse polyketide synthase gene clusters in microorganisms associated with the marine sponge Discodermia dissoluta. Applied Environmental Microbiology 71: 4840-4849. [ Links ]
57. Schloss, P. & J. Handelsman (2003). Biotecnological prospects from metagenomics. Current Opinion on Biotechnology 14: 303-310. [ Links ]
58. Schlotelburg, C., C. Wintzingerode, R. Hauck, F. Wintzingerode, W. Hegemann & U. Gobel (2002). Microbial structure of an anaerobic bioreactor population that continuously dechlorinates 1,2-dichloropropane. FEMS Microbiology Ecology 39: 229-237. [ Links ]
59. Sogin, M.L., H.G. Morrison, J.A. Huber, D.M. Welch, S.M. Huse, P.R. Neal, J.M. Arrieta & G.J. Herndl (2006). Microbial diversity in the deep sea and the underexplored "rare biosphere". Proceedings of the National Academy of Sciences USA 103: 12115-12120. [ Links ]
60. Sunnucks, P., A. Wilson, L. Beheregaray, K. Zenger, J. French & A. Taylor (2000). SSCP is not so difficult: the application and utility of single-stranded conformation polymorphism in evolutionary biology and molecular ecology. Molecular Ecology 9: 1699-1710. [ Links ]
61. Torsvik, V., J. Goksoyr & F. Daae (1990). High diversity in DNA of soil bacteria. Applied Environmental Microbiology 56: 782-787. [ Links ]
62. Turnbaugh, P.J., M. Hamady, T. Yatsunenko, B.L. Cantarel, A. Duncan, R.E. Ley, M.L. Sogin, W.J. Jones, B.A. Roe, J.P. Affourtit, M. Egholm, B. Henrissat, A.C. Heath, R. Knight & J.I. Gordon (2008). A core gut microbiome in obese and lean twins. Nature 457: 480-484. [ Links ]
63. Venter, J.C., K. Remington, J.F. Heidelberg, A.L. Halpern, D. Rusch, J.A. Eisen, D. Wu, I. Paulsen, K.E. Nelson, W. Nelson, D.E. Fouts, S. Levy, A.H. Knap, M.W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tillson, C. Pfannkoch, Y.H. Rogers & H.O. Smith (2004). Environmental Genome Shotgun Sequencing of the Sargasso Sea. Science 304: 66-74. [ Links ]
64. Wilson, I. (1997). Inhibition and facilitation of nucleic acid amplification. Applied Environmental Microbiology 63: 3741-3751. [ Links ]
65. Woese, C.R., O. Kandler & M.L. Wheelis (1990). Towards a Natural System of Organisms: Proposal for the Domains Archaea, Bacteria, and Eucarya. Proceedings of the National Academy of Sciences USA 87: 4576-4579. [ Links ]
66. Woese, C. (1998) The universal ancestor. Proceedings of the National Academy of Sciences USA 95: 6854-6859. [ Links ]
67. Yooseph, S., G. Sutton, D.B. Rusch, A.L. Halpern, S.J. Williamson, K. Remington, J.A. Eisen, K.B. Heidelberg, G. Manning, W. Li, L. Jaroszewski, P. Cieplak, C.S. Miller, H. Li, S.T. Mashiyama, M.P. Joachimiak, C. vanBelle, J.M. Chandonia, D.A. Soergel, Y. Zhai, K. Natarajan, S. Lee, B.J. Raphael, V. Bafna, R. Friedman, S.E. Brenner, A. Godzik, D. Eisenberg, J.E. Dixon, S.S. Taylor, R.L. Strausberg, M. Frazier & J.C. Venter (2007). The Sorcerer II Global Ocean Sampling expedition: Expanding the universe of protein families. PLoS Biology 5: 1-35. [ Links ]
68. Yun, J., S. Kang, S. Park, H. Yoon, M. Kim, S. Heu & S. Ryu (2004). Characterization of a novel amylolytic enzyme encoded by a gene from a soil-derived metagenomic library. Applied Environmental Microbiology 70: 7229-7235. [ Links ]

