










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Ciencia y Tecnología
versión On-line ISSN 1851-7587
Rev. cienc. tecnol. no.14 Posadas jul./dic. 2010
BIOQUÍMICA-FARMACIA
Estudio de las mutaciones CD39 e IVS1-110 causantes de β-talasemia mediante ARMS-PCR.
Study of CD39 and D/S1-110 mutations causing beta thalassemia by ARMS-PCR
Karina B. Acosta1, Mario A. Riera1, Nadia Labandera1, Pedro D. Zapata1, Zulema Galeano1.
1. Cátedra de Bioquímica Clínica I - Laboratorio de Biotecnología Molecular. Módulo de Bioquímica y Farmacia, Facultad de Ciencias Exactas Químicas y Naturales, UNaM. Av. Mariano Moreno 1375 (3300) - Posadas, Misiones, Argentina. (bcmb@fceqyn.unam.edu.ar)
• Karina Beatriz Acosta1 Es Licenciada en Genética. Se desempeña como Auxiliar Ad-Honorem de la Cátedra de Biología Celular de la Carrera de Licenciatura en Genética desde el año 2009. Realiza tareas de extensión y servicio en el Laboratorio de Biotecnología Molecular. Posee presentaciones a congreso.
• Mario Alejandro Riera1 Es alumno avanzado de la carrera de Bioquímica. Se desempeña como Auxiliar Alumno de la Cátedra de Biología Molecular y Genética desde el año 2006. Participa del Proyecto PICTO 37029 financiado por la ANPCyT, habiendo participado además del proyecto financiado por la ANPCyT (PICT 05-15058). Realiza tareas de extensión y servicio en el Laboratorio de Biotecnología Molecular. Posee publicaciones en revistas indexadas y presentaciones a congreso.
• Nadia Labandera1 Bioquímica Jefe de Trabajos Prácticos Regular en las asignaturas Bioquímica Clínica I y II de la carrera de Bioquímica de la Facultad de Ciencias Exactas Químicas y Naturales e investigadora categoría 5 en el Sistema Nacional de Incentivos Docentes - Investigadores.
• Pedro Darío Zapata1 Es Doctor por la Universidad de Alcalá de Henares. Se desempeña actualmente como Profesor Regular Adjunto en las Cátedras de Biología Celular y Molecular (Bioquímica), Genética Molecular, Biología Celular (Lic. en Genética) y Biotecnología Molecular (Ing. Química, Bioquímica y Farmacia). Posee actualmente la Categoría I en el Sistema Nacional de Incentivos a los Docentes-Investigadores. En el área de Biomedicina ha sido beneficiario de subsidios de la ANPCyT: PICT 05-15058 y PICTO 37029. Dirige auxiliares, becarios de grado y posgrado. Posee publicaciones en revistas indexadas nacionales e internacionales y presentaciones a congresos nacionales e internacionales.
• Zulema Galeano1 Bioquímica Profesora Regular Adjunto a cargo de las asignaturas Bioquímica Clínica I y II de la carrera de Bioquímica de la Facultad de Ciencias Exactas Químicas y Naturales e investigadora categoría 4 en el Sistema Nacional de Incentivos Docentes - Investigadores.
Resumen
Las β-talasemias obedecen a una disminución en la síntesis de cadenas de β-globina, y son causadas por diferentes mutaciones en el gen de β-globina. La detección molecular de estas mutaciones, permite determinar el genotipo de β-talasemia y así establecer con precisión el diagnóstico y el tratamiento a seguir. El objetivo del presente trabajo fue estudiar las dos mutaciones de hallazgo más frecuente en la población argentina, CD39 e IVS1-110 del gen de β-globina, mediante la técnica de ARMS-PCR. Del total de pacientes analizados (50), 3 resultaron positivos para la mutación CD39, correspondiente a un 6 % de los casos, mientras que no fueron detectados pacientes portadores de la mutación IVS1-110. Este estudio aporta nuevos datos sobre la frecuencia de los genotipos (β/βCD39 y β/βIVS1-110) responsables de β-talasemia heterocigota en nuestra región.
Palabras clave: β-talasemia; Mutaciones; Genotipo; Diagnóstico; ARMS-PCR.
Abstract
β-thalassemias derive from a decrease in the synthesis of β-globin chains, being caused by different mutations in the β-globin gene. The molecular detection of these mutations allows the β-thalassemic genotype determination and therefore, a precise diagnosis and the underlying treatment. The aim of this study was to study the two most common finding mutations in the Argentinean population, CD39 and IVS1-110 mutations in the β-globin gene, by the ARMS-PCR technique. In 50 patients analyzed, three were positive for the CD39 mutation, corresponding to 6 % of the cases, while no patients with the IVS1-110 mutation were detected. This study provides new data on the frequency of genotypes (β/βCD39 and p / βIVS1-110) responsible for β-thalassemia heterozygote in our region.
Key words: β-thalassemias; Mutations; Genotype; Diagnosis; ARMS-PCR.
Introducción
Las talasemias, descriptas originalmente por Cooley y Lee (1925), se definen como un grupo heterogéneo de trastornos genéticos en la síntesis de la hemoglobina (Hb), caracterizados por una reducción en el grado de producción de una o más cadenas de globina, lo cual lleva a un desbalance en la síntesis de las mismas, una concentración disminuida de Hb en los eritrocitos y finalmente anemia [1, 2].
Las β-talasemias, se deben a una disminución en la síntesis de cadenas de beta globina (β-globina), y son causadas por diferentes mutaciones en este gen, que incluyen desde la región promotora hasta la señal de poliadenilación. La intensidad del déficit depende del grado de alteración genética y puede variar desde una síntesis deficiente o parcial (talasemia β+) hasta una ausencia total de síntesis (talasemia β0). Clínicamente, se pueden clasificar en tres grandes grupos: la β-talasemia mayor, que comprende a los genotipos homocigotos (β0/β0 y β+/β+) o dobles heterocigotos (β0/β+), y corresponde a las formas de mayor expresividad clínica.
Por otro lado se encuentran la β-talasemia intermedia, con expresividad clínica variable; y la β-talasemia menor que agrupa a los genotipos heterocigotos (β+/P o β0/P). Esta última corresponde a la forma más frecuente en nuestro país, presentando expresividad clínica poco manifiesta o ausente, con disminución en los parámetros hemáticos de VCM (Volumen Corpuscular Medio) y HCM (Hemoglobina Corpuscular Media), respecto a sus valores normales (82 fl y 27 pg, respectivamente) [3, 4].
Aunque se han identificado más de 200 mutaciones causantes de β-talasemia en todo el mundo; cada población tiene su propio repertorio de mutaciones, aunque un número limitado constituyen las más comunes [5]. Particularmente, en la Región Mediterránea se han identificado más de 30 mutaciones β-talasémicas, pero solamente 8 de ellas son las más frecuentes. Estas 8 mutaciones representan el 95 % de los genes afectados, demostrados en poblaciones de Argentina, donde las dos más frecuentes, una β0 y la otra β+, abarcan el 68,9 % de los casos, un 45,7 % para la mutación del codón 39 (β0) y un 23,2 % para el intrón 1, en el nucleótido 110 (β+) [6, 7, 8].
Las formas heterocigotas (β/β0, β/β+) generalmente originan un cuadro clínico asintomático [9], por lo que el diagnóstico suele ser casi siempre casual por algún examen hematológico de rutina. Este examen incluye, entre otras, la determinación de parámetros hemáticos (VCM, HCM) y cuantificación de Hbs; pero en prácticamente todos los casos, se basa en la dosificación de la HbA2 (aumentada) y en un estudio familiar [4]. Sin embargo, los estudios hematológicos presentan ciertas limitaciones, ya que existen casos de β-talasemia heterocigota en los que la fracción de HbA2 no se incrementa [10] y tampoco permiten la distinción entre los tipos β0 y β+ de talasemia [11, 12]. Por lo tanto, si bien los estudios hematológicos son muy útiles para la detección de β-talasemia, no permiten precisar con exactitud el defecto causante del padecimiento, situación que muchas veces puede conducir a un diagnóstico erróneo (ferropenia con características similares) y a un tratamiento equivocado, que puede agravar la condición del paciente (administración de hierro a un paciente talasémico) [13]. Esta situación evidencia la necesidad de implementar técnicas de Biología Molecular, complementarias a los métodos hematológicos, para determinar el genotipo de β-talasemia y establecer con precisión el diagnóstico y el tratamiento a seguir [10, 13].
La aplicación de métodos analíticos basados en la técnica de PCR [14], han facilitado la detección de los genotipos causantes de β-talasemia [15], ya que permiten discriminar entre alelos normales y mutantes que difieren en una sola base, siendo altamente eficientes y de bajo costo [16].
El método Amplificación Refractaria de Sistemas de Mutaciones (ARMS-PCR) es una modificación de la técnica de PCR [14], descrito por primera vez por Newton y cols., en 1989 [17]; y utilizada para la detección de mutaciones puntuales causantes de β-talasemia [18]. Esta técnica permite la amplificación enzimática de alelos específicos, mediante el uso de cebadores que están diseñados para discriminar entre secuencias que difieren en una única base. Además, utiliza cebadores control que amplifican otra región del gen de P globina, cercana a la mutación que será detectada, actuando como control interno de amplificación asegurando la eficiencia de la PCR y evitando falsos negativos [17].
El objetivo del presente trabajo fue estudiar en una población de pacientes con diagnóstico presuntivo de β-talasemia heterocigota, las dos mutaciones más frecuentes en la población argentina, a través de la búsqueda de la mutación C→T en el codón 39 (CD 39; carácter β0) y G→A en el nucleótido 110 del intrón 1 (IVS1-110; carácter β+) del gen de P globina; con un método rápido, simple y de bajo costo, la ARMS-PCR o PCR con oligonucleótidos mutados.
Materiales y métodos
Muestras utilizadas
En este trabajo se analizaron 50 muestras de sangre entera de pacientes portadores de anemia crónica, no respondedores al tratamiento convencional con hierro y con diagnóstico presuntivo de β-talasemia heterocigota, derivados por médicos hematólogos de diferentes nosocomios de la ciudad de Posadas.
Los controles positivos fueron cedidos por la Dra. Margarita Bragós de la Cátedra y Servicio de Hematología de la Facultad Ciencias Bioquímicas y Farmacéuticas de la Universidad Nacional de Rosario.
Además de los datos hematológicos, que incluían parámetros de los eritrocitos y porcentaje de HbA2, se contaba con información sobre ascendencia geográfica de los pacientes.
Al tratarse de muestras provenientes de humanos, se solicitó el consentimiento informado de cada persona, y se protegió la identidad de los individuos mediante la codificación de las muestras, siguiendo las reglamentaciones éticas, legales y jurídicas establecidas en las normas bioéticas nacionales -Disposición ANMAT 5330/97- e internacionales -Código de Nüremberg, Declaración de Helsinski y sus modificaciones; así como también la Declaración Universal sobre Genoma Humano y Derechos Humanos aprobada por la Conferencia General de la UNESCO, del 11/11/1997.
Extracción de DNA
Las extracciones de DNA a partir de muestras de sangre entera se llevaron a cabo por el método de salting out, según el protocolo modificado de Miller y cols., 1988 [19, 20].
La integridad del DNA extraído se verificó en geles de agarosa al 1 %, teñidos con bromuro de etidio (10mg/ ml, Promega) y la semicuantificación del DNA se realizó por medio de la comparación de la intensidad de bandas con un patrón de concentración conocida (K562 10ng/μl, Promega). El análisis visual a UV proporcionó información relativa a concentraciones (ng/μl) y calidad de DNA extraído.
Estudio de las mutaciones p talasémicas
Para el estudio de las mutaciones CD39 e IVS1-110 se aplicó la técnica de ARMS-PCR, utilizando cebadores descriptos previamente en la bibliografía [18] y detallados en las Tablas 1 y 2.
Tabla 1. Secuencias de los cebadores alelo específicos.

Tabla 2. Secuencias de los cebadores del control interno de amplificación.

Las reacciones de PCR fueron realizadas en un volumen final de 50 μl conteniendo tampón PCR (KCl 1X), 800 uM de dNTPs, 2,5 mM de MgCl2, 2 U de Taq polimerasa (Fermentas) y 80 ng/μl de DNA. Las concentraciones de los cebadores del control interno y alelo específicos (OPERON Technologies, Alemania) fueron de 20 pmoles y 10 pmoles, respectivamente, para el estudio de la mutación CD39; y 20 pmoles de ambos cebadores para el estudio de la mutación IVS1-110. El ciclado aplicado consistió en una etapa inicial de 2 min a 93 °C, seguidos de 25 ciclos de amplificación conformado por un período de desnaturalización a 93 °C durante 1 min, un período de hibridación a 65 °C durante 1 min y de extensión a 72 °C durante 1 min 30 s, con un período final de extensión 3 min a 72 °C. Las reacciones de PCR fueron llevadas a cabo en un equipo termociclador MultiGENE II Thermal Cycler (TC 020A-230V).
Para verificar los resultados de la amplificación, las muestras fueron sometidas a electroforesis en geles de agarosa al 2 %, teñidos con bromuro de etidio (10mg/ml, Promega) y corridas a 100 V por 30 minutos con buffer TBE 0,5X y azul de bromofenol. Posteriormente, fueron visualizados en un transiluminador de luz UV (MUV21-312-220) y fotografiados con cámara digital ACER (CS-5531).
Resultados y Discusión
Perfil hematológico de la población analizada
El 100 % de los pacientes analizados (50) se caracterizó por la presencia de microcitosis e hipocromía con valores de VCM y HCM por debajo de sus valores normales (82 fl y 27 pg, respectivamente), y el 40 % (20) presentó niveles aumentados de HbA2 (valor de referencia 3,5 %); parámetros indicativos de presunción diagnóstica de β-talasemia heterocigota.
Estudio de las mutaciones OD39 e IVS1-110 causantes de β-talasemia por ARMS-por
El presente estudio constituye el primer registro sobre la frecuencia de los genotipos (β/βCD39 y β/ βIVS1-110) responsables de β-talasemia heterocigota en nuestra región.
Del total de pacientes analizados (n = 50), 3 resultaron positivos para la mutación CD39 (6 % de los casos), mientras que no fueron detectados pacientes portadores de la mutación IVS1-110.
En los individuos positivos para la mutación CD39, se observó la presencia de las bandas correspondientes al alelo normal y mutado, junto con las bandas del control interno de amplificación; mientras que en los casos en los que no se identificó ninguna de las dos mutaciones (CD39 e IVS1-110), se observó la banda del control interno, lo que garantizó que no se trataran de falsos negativos por fallas en la PCR.
La Figura 1 muestra el perfil de bandas obtenidas en el estudio de la mutación CD39. La presencia de una banda de 436 pb en el ensayo mutado, además de la banda del control interno de 862 pb, demostró que tres de los pacientes analizados eran portadores de la mutación en cuestión.
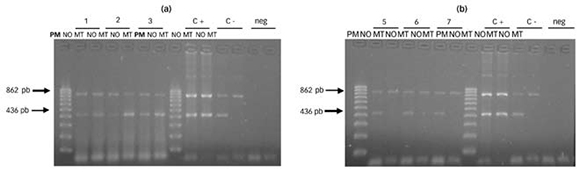
Figura 1. Perii! de bandas obtenidas en el estudio de la mutación CD39. Electroforesis en geles de agarosa al 2 %. A). Se observan los ensayos normal (NO) y mutado (MT) de 3 pacientes positivos para la mutación CD39. B). Se observan los ensayos normal y mutado de 3 pacientes negativos para la mutación CD39. En ambos geles se observan también los ensayos normal y mutado del control positivo (C+) y negativo (c-) de alelos; los controles negativos de PCR (neg); y el marcador de peso molecular de I00pb (pm). Se indican las bandas de control interno (862 pb) y la banda correspondiente al alelo normal o mutado (436 pb).
En ambos geles se observan también los ensayos normal y mutado del control positivo (C+) y negativo (C-) de alelos; los controles negativos de PCR (neg); y el marcador 32 de peso molecular de 100 pb (PM). Se indican las bandas de control interno (862 pb) y la banda correspondiente al alelo normal o mutado (436 pb).
La Figura 2 muestra el perfil de bandas obtenidas en el estudio de la mutación IVS1-110, en el que no se observó la presencia de la banda de 390 pb correspondiente al alelo mutado, lo que indicó ausencia de esta mutación en los pacientes analizados.
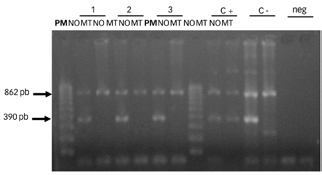
Figura 2. Perfil de bandas obtenidas en el estudio de la mutación IVS1-110. Electroforesis en geles de agarosa al 2 %. Se observan los ensayos normal y mutado de 3 pacientes negativos para la mutación CD39. Se observan también los ensayos normal y mutado del control positivo (C+) y negativo (c-) de alelos; los controles negativos de PCR (neg); y el marcador de peso molecular de I00pb (pm). Se indican las bandas de control interno (862 pb) y la banda correspondiente al alelo normal o mutado (390 pb).
Los tres pacientes positivos para la mutación CD39 son de ascendencia italiana, italiana/española, y alemana/ polaca. En el análisis de los datos hematológicos de estos pacientes se observó, en dos de ellos, valores de VCM, HCM y HbA2 que concuerdan con los criterios establecidos en el diagnóstico hematológico de β-talasemia heretocigota, presentando microcitosis, hipocromía y niveles de HbA2 superiores a 3,5 %. Sin embargo, uno de los pacientes portadores de esta mutación, no cursaba con elevación de HbA2, presentando un valor inferior a 3,5 %, que podría deberse a una deficiencia concomitante de hierro que impide el incremento de esta fracción de Hb [10]. En este último caso, la aplicación de la técnica de ARMS-PCR fue fundamental para establecer el diagnóstico concreto de β-talasemia heterocigota, ya que el valor de HbA2 no permitía definirlo por el método convencional.
Los pacientes portadores de éstas (hetrerocigotos β/βCD39 y β/βIVS1-110) y otras mutaciones β-talasémicas, son generalmente salvo complicaciones asintomáticos. Sin embargo, en estado homocigótico producen cuadros clínicos de mayor gravedad (Anemia de Cooley). De aquí la importancia de detectar a los individuos portadores, y en especial, aquellas parejas con probabilidades de concebir hijos con talasemia mayor, teniendo en cuenta las complicaciones que conlleva la herencia de dicha condición. El diagnóstico molecular de β-talasemia, puede ser útil para brindar el asesoramiento genético entre parejas portadoras [21].
La técnica de ARMS-PCR aplicada en este estudio, reúne los requisitos necesarios de los métodos de diagnóstico: alta especificidad, reproducibilidad y bajo costo [22]. Por lo tanto constituye un método eficaz para el diagnóstico de β-talasemia en pacientes sin posibilidad de estudio familiar, debido a la falta de uno de los padres. Esta técnica no sólo permite detectar la mutación causante del padecimiento, sino también determinar si se encuentran en estado homocigota o heterocigota, siendo particularmente útil su aplicación en estudios poblacionales, a fin de poder identificar la frecuencia real de la β-talasemia menor [23].
El diagnóstico de β-talasemia menor no se realiza a menudo por muchos años. Los portadores pueden identificarse la mayoría de las veces mediante un examen hematológico convencional. Sin embargo, cuando los niveles de HbA2 se encuentran dentro de los valores normales (por ferropenia asociada), no es posible realizar el diagnóstico preciso de β-talasemia mediante el método convencional. Esta situación se resuelve con técnicas como la ARMS-PCR, que a nivel molecular pueden determinar los defectos causantes del padecimiento en una población, sin depender de los niveles de HbA2[23]. Establecer el diagnóstico preciso, es fundamental para evitar que el paciente realice continuas investigaciones y tratamientos innecesarios con hierro, generalmente contraproducentes; y por otra parte, para proceder al asesoramiento genético pertinente.
Conclusiones
Se logró detectar la presencia de la mutación CD 39 (C→T) de carácter ß° en las muestras de tres pacientes, logrando caracterizarlos genotípicamente y confirmando el diagnóstico de ß-talasemia heterocigota; mientras que no fueron detectados pacientes portadores de la mutación IVS1-110.
Los resultados negativos obtenidos en este estudio, no son excluyentes de la condición talasémica, ya que estos pacientes podrían presentar ß-talasemia heterocigota debida a otra mutación diferente a las estudiadas en este trabajo.
Referencias bibliográficas
1. Natta C., Banks J., Niazi G., Marks P.A. y Bank A. Decreased globin mRNA activity o homozygous and heterozygous ß-thalassemia. Nature, 244: p. 280-283. 1973. [ Links ]
2. Bank A. The thalassemia syndromes. Blood, 51: p. 369-374. 1978. [ Links ]
3. Naoum P.C. Hemoglobinopatias e talassemias. Säo Paulo: Sarvier, pp. 169. 1997. [ Links ]
4. Malcorra J.J. Hemoglobinopatias y Talasemias. BSCP Can Ped, 25(2): p. 265-277. 2001. [ Links ]
5. Weatherall D.J., Clegg B.J., Higgs D.R. y Wood W.G. The hemoglobinopathies. En: Sariver C., Beaudet A. L., Sly W., Valle D. y Childs B. The Metabolic and Molecular Bases of Inherited Disease. 8a ed. Nueva York: McGraw-Hill, pp. 4571-4636. 2001. [ Links ]
6. Roldan R., Gutierrez M., Cygler A., Bonduel M., Sciuccati G. y Torres A.F. Molecular characterization of ß-thalassemia genes in an Argentine population. American Journal of Hematology, 54: p. 179-182. 1997. [ Links ]
7. Bragós I.M., Noguera N.I., Morisoli L. y Milani A.G. Most frequent mutations of ß-thalassemia in Rosario, Argentina. Haematologica, 85(1): p. 101-102. 2000. [ Links ]
8. Rossetti L., Targovnik H. y Varela V. The molecular basis of ß-thalassemia in Argentina. Influence of the pattern of inmigration from the Mediterranean Basis. Haemotologica, 89(6): p. 746-747. 2004. [ Links ]
9. Bunn H.F., Forget B.G. y Ranney, H.M. Hemoglobinopathies. En su: Major Problems in Internal Medicine. Philadelphia, USA: W. B. Saunders Co., pp. 70-73. 1976. [ Links ]
10. Ruiz-Reyes G. Los síndromes talasémicos no son infrecuentes en la población mexicana y se subdiagnostican y confunden con deficiencias de hierro. Medicina Universitaria, 1(2): p. 67-73. 1999. [ Links ]
11. Fessas Ph y Loukopoulos D. Las talasemias beta. En su: Clínica Hematológica (Hemoglobinas Anormales). (s.l.): (s.n.), pp. 199-224. 1976. [ Links ]
12. Sáenz G.F., Elizondo J. y Páez C.A. Hallazgo del gen beta0-talasémico (supresor) en Costa Rica V. Síndrome de heterocigosis doble S/beta0-tal. Sangre, 23:196-201. 1978. [ Links ]
13. Lynes Decle Mdc. Frecuencia de siete mutaciones conocidas del gen de la beta globina en pacientes mestizos mexicanos con talasemia beta o delta beta. Tesis de Maestría en Biomedicina Clínica. Departamento de Ciencias Químico-Biológicas, Escuela de Ingeniería y Ciencias. Universidad de las Américas Puebla, 2007. [ Links ]
14. Mullis K.B., Faloona F., Scharf S.J., Saiki R., Horn G. y Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp Quant Biol. 51: p. 263-273. 1986. [ Links ]
15. Kanavakis E., Traeger-Synodinos J., Vrettou C., Maragoudaki E., Tzetis M. y Kattamis C. Prenatal diagnosis of the thalassaemia syndromes by rapid DNA analytical methods. Molecular Human Reproduction, 3(6): p. 523-528. 1997. [ Links ]
16. Benz E.J. Jr. The thalassemia syndromes: lessons from molecular medicines index case. Trans Am Climatol Assoc, 107: p. 20-36. 1995. [ Links ]
17. Newton C.R., Graham A., Heptinstall L.E., Powell S.J., Summers C., Kalsheker N., Smith J.M., Markham A.F. Analysis of any mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Research, 17(7): p. 2503-2516. 1989. [ Links ]
18. Old J.M., Varawalla N.Y. y Weatherall D.J. Rapid detection and prenatal diagnosis of ß thalassaemia: studies in Indian and Cyproit populations in the UK. Lancet, 336: p. 834-37. 1990. [ Links ]
19. Miller S.A., Dykes D.D. y Polesky H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 16 (3): p. 1215. 1988. [ Links ]
20. Cariaga-Martinez, A.E. y Zapata, P.D. El laboratorio de Biología Molecular: una guía práctica. Edición ampliada. Editorial Universitaria de Misiones, Serie Cátedra, 2007. [ Links ]
21. Sáenz G.F., Chaves M., Montero A.G., y Jiménez J. Síndromes de beta talasemia menor o heterocigota II. Aspectos analítico-diagnósticos. Rev. Cost. Cienc. Méd., 9(1): p. 83-91. 1988. [ Links ]
22. Eng B., Walker L., Nakamura L.M., Hoppe C., Azzimi M., Lee H. y Waye J.S. Three new beta-globin gene promoter mutations identified through newborn screening. Hemoglobin, 31: p. 129-134. 2007. [ Links ]
23. Bravo M., Salazar R., Arends A., Alvarez M., Velázquez D., Guevara J.M. y Castillo O. Detección de ß talasemia mediante la técnica de Amplificación Refractaria de Sistemas de Mutaciones (ARMS-PCR). Invest Clin, 40(3): p. 203-213. 1999. [ Links ]
Recibido: 05/08/10.
Aprobado: 07/04/11
