










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista americana de medicina respiratoria
On-line version ISSN 1852-236X
Rev. am. med. respir. vol.14 no.2 CABA June 2014
ARTÍCULOS ORIGINALES
Hipertensión arterial pulmonar. Registro de un centro de referencia en Argentina
Autores: María L. Talavera1, Jorge O. Cáneva1, 2, Liliana E. Favaloro1, Francisco Klein1, Roberto P. Boughen1, Gerardo E. Bozovich3, Juan M. Ossés2, 4, Roberto R. Favaloro4, Alejandro M. Bertolotti4
1Equipo de Hipertensión Pulmonar del Hospital Universitario Fundación Favaloro. Buenos Aires, Argentina
2Servicio de Neumonología del Hospital Universitario Fundación Favaloro. Buenos Aires, Argentina
3Director médico del Hospital Universitario Fundación Favaloro. Buenos Aires, Argentina
4División de Trasplante Intratorácico del Hospital Universitario Fundación Favaloro Buenos Aires, Argentina
Correspondencia: María Luján Talavera Domicilio postal: Av Belgrano 1782, primer piso. (C1093AAS) Buenos aires, Argentina. Tel./fax: 5411 43781247 E-mail: marialujant@gmail.com
Recibido: 26.02.2014
Aceptado: 23.04.2014
Resumen
Introducción y objetivos: En Argentina, no se conocen datos de pacientes con hipertensión pulmonar (HP) con diagnóstico de certeza. El propósito del presente registro fue conocer las características de nuestra población de pacientes con HP del Grupo I de la clasificación de Dana Point 2008, hipertensión arterial pulmonar (HAP), estudiar la supervivencia y las variables asociadas a mayor mortalidad.
Métodos: Estudio de cohorte de 134 pacientes admitidos por primera vez en el consultorio de HP de nuestra institución en el período comprendido entre enero/2004 y marzo/2012. Se excluyeron 9 pacientes; total analizado: 125 pacientes.
Resultados: Edad promedio: 34 ± 15,7 años y la relación mujer/hombre: 3,8/1. Las etiologías más prevalentes fueron la HAP idiopática (HAPI) (61, 48.8%), asociadas a cardiopatías congénitas (35, 28%) y a colagenopatías (18, 14.4%).
La supervivencia libre de trasplante o muerte fue de 63.2%; a 12, 24 y 36 meses la supervivencia fue del 94% (IC95% 88-97), 90% (IC95% 83-94) y 83% (IC95% 75-89), respectivamente.
Las variables basales asociadas a mayor mortalidad y/o trasplante fueron la disnea clase funcional III/IV (OR 3.6 IC95% 1.5-8.9, p < 0.01), la distancia recorrida en la prueba de la caminata de 6 minutos menor de 380 m (OR 2.7 IC95% 1.1-6.5 p = 0.023) y el derrame pericárdico (OR 4.2 IC95% 1.3-14.6 p = 0.021).
Conclusiones: Esta serie muestra las características y supervivencia actuales de pacientes con HP en un centro de referencia en Argentina. Variables de fácil obtención y reproducción permitieron estratificar subgrupos de mayor riesgo de muerte y/o trasplante en el seguimiento.
Palabras clave: Hipertensión pulmonar; Hipertensión arterial pulmonar; Supervivencia.
Abstract
Pulmonary Arterial Hypertension. A Registry at a Reference Centre in Argentina
Introduction and objectives: There are no data on outcome of patients with pulmonary hypertension (PH) in Argentina. The objective of this registry was to assess the characteristics of patients with PH belonging to the Group I Dana Point's 2008 classification, pulmonary arterial hypertension (PAH), and to identify variables associated with outcomes.
Methods: One hundred and thirty four patients were analyzed in a consecutive sequence. After excluding nine patients, 125 patients were analyzed.
Results: The patients mean age was 34 years (SD±15,7) and the female/male ratio was 3.8/1. The most prevalent aetiologies were: idiopathic PAH (IHAP) (61, 48.8%), PH associated to congenital heart disease (35, 28%) and PH associated to connective tissue disease (18, 14.4%).
Overall survival, excluding transplantation, was 63.2%; at 12, 24 and 36 months survival was 94% (CI 95% 88-97), 90% (CI 95% 83-94) and 83% (CI 75-89), respectively.
Baseline variables associated to increased mortality or need for transplantation were functional class III/IV (NYHA) (OR 3.6 CI 95% 1.5-8.9, p <0.01), 6 minute-walk test distance shorter than 380 m (OR 2.7 CI 95% 1.1-6.5 p = 0.023) and pericardial effusion (OR 4.2 CI 95% 1.3-14.6 p = 0.021).
Conclusion: This series shows characteristics and survival of patients with pulmonary arterial hypertension at a reference centre in Argentina. Some readily available variables allowed us to stratify subgroups at a higher risk of death and/or transplantation during the follow up.
Key words: Pulmonary hypertension; Pulmonary arterial hypertension; Survival.
Abreviaturas:
HP: hipertensión pulmonar
HAP: hipertensión arterial pulmonar
HAPI: hipertensión pulmonar idiopática
CF: clase funcional
CCD: cateterismo cardíaco derecho
PC6M: prueba de la caminata de 6 minutos
PAPm: presión de la arteria pulmonar media
PCEP: presión capilar de enclavamiento pulmonar
Abbreviations:
HP: pulmonary hypertension
PAH: pulmonary arterial hypertension
IPAH: idiopathic pulmonary arterial hypertension
FC: functional class
RHC: right heart catheterization
6MWT: 6-minute walking test
mPAP: mean pulmonary arterial pressure
PAWP: pulmonary arterial wedge pressure
Introducción
La hipertensión arterial pulmonar (HAP) es una enfermedad caracterizada por un incremento progresivo de la resistencia vascular pulmonar que conduce al fallo ventricular derecho y muerte, e incluye sus formas idiopática (HAPI) y asociadas a otras condiciones como las colagenopatías, cardiopatías congénitas o virus de la inmunodeficiencia adquirida, entre otras1, 2.
En las últimas décadas, se han producido grandes avances en la comprensión de la fisiopatología de la enfermedad y esto derivó en el desarrollo de nuevos blancos terapéuticos3. A pesar de estos progresos, la enfermedad aún no posee una cura definitiva.
En 1981, el Instituto Nacional de la Salud de Estados Unidos llevó a cabo el primer registro de pacientes con HAPI, entonces denominada hipertensión pulmonar (HP) primaria, en donde estableció una supervivencia media de 2,8 años con tasas de supervivencia de 68%, 48% y 34% a 1, 3 y 5 años, respectivamente4.
No obstante, en los últimos 15 años se ha avanzado de manera considerable en el diagnóstico y tratamiento de esta enfermedad; actualmente se realiza una evaluación sistemática y más objetiva como la medición de parámetros hemodinámicos mediante el cateterismo cardíaco derecho (CCD) en forma rutinaria, valoración de la capacidad de ejercicio mediante la prueba de caminata de los 6 minutos (PC6M) y la determinación de marcadores bioquímicos (péptidos natriuréticos y troponinas). A su vez, se han incorporado al tratamiento nuevas drogas como los derivados prostanoides, los antagonistas de receptores de endotelina 1 y los inhibidores de la fosfodiesterasa 5. Así, los últimos registros han reportado una supervivencia significativamente superior a la observada en la década de los '80. Los últimos registros llevados a cabo en Francia, Estados Unidos y Reino Unido muestran una supervivencia al año del 88%, 91% y 92,7%, respectivamente5-7.
El presente registro se realizó con el objetivo de conocer las características, el manejo diagnóstico y terapéutico, y la supervivencia de pacientes con HAP en un centro de referencia en Argentina. Un interrogante fue conocer si las diferencias étnicas, culturales, ambientales y socioeconómicas de esta región, comparada con Europa o Norteamérica, podrían influir en la evolución de la enfermedad.
Material y métodos
Se efectuó un análisis post hoc a partir de datos colectados en prospectiva y consecutiva de 134 pacientes con diagnóstico de HAP, Grupo I según la clasificación actual de Dana Point del 2008, en seguimiento por el Grupo de Hipertensión Pulmonar de esta institución desde enero de 2004 a marzo de 20128. Nueve pacientes fueron excluidos por pérdida de seguimiento.
Se definió HAP a la presencia de una presión en arteria pulmonar media (PAPm) ≥ 25 mmHg y una presión capilar de enclavamiento pulmonar (PCEP) ≤ 15mmHg medidas por CCD9. Al momento de su admisión, los pacientes fueron estudiados mediante CCD, ecocardiograma Doppler con contraste (solución salina agitada), laboratorio de sangre: hemograma, coagulación, función renal y hepática, parámetros de colagenopatía (factor reumatoideo, anticuerpos antinucleares, anti DNA, anti Scl70), serologías para el virus de la inmunodeficiencia adquirida y de la hepatitis B y C, función tiroidea, laboratorio de función pulmonar, PC6M y centellograma pulmonar ventilación-perfusión. En caso de que este último hubiera resultado de probabilidad alta/intermedia para tromboembolismo pulmonar, se procedió a una angiografía pulmonar en el mismo procedimiento del CCD para evaluar la presencia de HP tromboembólica crónica y, ante este diagnóstico, se excluyeron del análisis por pertenecer al Grupo 4 de la clasificación de Dana Point.
La fecha de diagnóstico de HP se estableció el día que se efectuó el CCD. Se excluyeron aquellos con una PCEP>15 mmHg por considerarlos pertenecientes al Grupo 2 (Dana Point).
Los pacientes con patología respiratoria clínicamente significativa, definida por capacidad vital forzada y/o un volumen espiratorio forzado en el primer segundo <60% de sus valores de referencia, fueron excluidos porque se asumió que este grupo puede presentar HP secundaria a su patología respiratoria (Grupo III, Dana Point)8.
La prueba aguda de reactividad pulmonar fue efectuada durante el CCD con iloprost inhalado en el 95% de los casos y con óxido nítrico inhalado en el 5% restante. El resultado de la misma fue definido como positivo cuando se produjo una reducción de la PAPm ≥ 10 mmHg y un valor absoluto final de la prueba ≤ 40 mmHg, con un índice cardíaco (IC) sin variaciones durante la prueba10.
Se definió caso incidental a aquel en que los síntomas hubiesen comenzado en los 6 meses previos al CCD y caso prevalente al que presentaba síntomas con más meses de evolución. Se definió CF avanzada a las categorías III y IV de la NYHA11. Se definió deterioro significativo de la función sistólica del ventrículo derecho a la presencia de deterioro al menos moderado de la misma por ecocardiograma Doppler por visualización directa del ventrículo derecho por operadores con experiencia en esta patología.
El presente manuscrito fue revisado y aprobado por el Comité de Bioética de la Institución.
Análisis estadístico
Las variables cualitativas fueron expresadas a través de proporciones y comparadas por medio de la prueba de Chi2 o exacta de Fisher, según correspondiera. Las variables continuas se describieron a través de media y desvío estándar, o mediana e intervalo intercuartilo 25-75 y fueron evaluadas a través de la prueba de t o Wilcoxon, según su distribución. En todos los casos se aplicó un valor de alfa de 0,05. Identificadas las variables de confusión, realizamos un análisis de regresión logística con eliminación retrógrada para establecer asociaciones ajustadas.
Construimos funciones de supervivencia por método de Kaplan y Meier a 12, 24 y 36 meses, comparando grupos por medio de la prueba "log rank". El análisis estadístico se llevó a cabo mediante STATA ver 11.2 (STATA Corp, College Station, Texas, EE UU).
Resultados
Población del estudio
Se realizó el análisis de los pacientes admitidos por primera vez en el consultorio de HP de nuestra institución en el período comprendido entre enero/2004 y marzo/2012. Fueron ingresados en forma consecutiva 134 pacientes. Nueve pacientes fueron excluidos por falta de seguimiento de tal modo que el total analizado fue de 125.
La mediana de seguimiento fue de 39.1 (IIC25- 75 21.2-61.5) meses. Las características basales de la población se resumen en la Tabla 1. Hubo predominancia de mujeres (99, 79.2%) con una relación mujer/hombre de 3,8/1. La edad promedio fue de 34 años (DS ± 15.7) y, del total de la población, hubo 16 pacientes (12.8%) < 18 años y 11 > 60 años (8.8%). El 26.4% (33 pacientes) representaron casos incidentales. De los 35 pacientes con cardiopatías congénitas, 28 tenían síndrome de Eisenmenger (15 presentaban defectos pretricuspídeos y 13 postricuspídeos) y 7 diagnóstico previo de comunicación interauricular cerrada.
Tabla 1. Características de la población

CF: clase funcional; ECG: electrocardiograma; HVD: hipertrofia ventricular derecha; FSVD: función sistólica del ventrículo derecho; PM6M: prueba de caminata de 6 minutos;IC: insuficiencia cardíaca
En relación a las etiologías, la HAPI fue la más frecuente (61, 48.8%) seguida por las asociadas a cardiopatías congénitas (35, 28%) y a colagenopatías (18, 14.4%); 8 presentaron HAP portopulmonar (6.4%), 2 HAP familiar (1.6%) y 1 asociada al virus de la inmunodeficiencia adquirida (0.8%). Al momento del diagnóstico, la mediana de evolución desde el inicio de los síntomas y el diagnóstico de la enfermedad fue de 16.2 meses (IIC25-75 7.5-37.9). El 58.4% se encontraba en CF avanzada (CF I: 8%, CF II: 33.6%, CF III: 48%, CF IV: 10.4%).
El electrocardiograma mostró ritmo sinusal en la mayoría de los pacientes (98.4%), sólo 3 mostraron trastornos del ritmo (2 fibrilación auricular, 1 aleteo auricular). En el ecocardiograma, 71 pacientes mostraban deterioro significativo de la función sistólica del ventrículo derecho (58,7%, 71/121 pacientes) y 18 tenían derrame pericárdico (14,9%, 18/121 pacientes).
Al momento del diagnóstico, se contó con una valoración de la capacidad de ejercicio en el 89% de la población. El método utilizado fue la PC6M, la cual fue anormal en la mayoría: 54.7% y 54.9% del valor de referencia para mujeres y hombres, respectivamente12, 13, con una mediana de 360 m (IIC25-75 247-432 m).
Los datos del CCD se muestran en la Tabla 2. La prueba aguda de vasorreactividad pulmonar fue realizada en 89 pacientes (71.2%). El resultado del estudio fue considerado positivo en el 9% (8 pacientes) de los casos, y principalmente limitado a la población con HAPI.
Tabla 2. Cateterismo cardíaco derecho

PAPs: presión arterial pulmonar sistólica; PAPm: presión arterial pulmonar media; PADm: presión auricular derecha media; RVP: resistencia vascular pulmonar; IC: índice cardíaco; PCP: presión capilar pulmonar; PAVRP: prueba aguda de vasorreactividad pulmonar; Respuesta: respuesta positiva a la PAVRP.
Al momento del análisis, el 81.6% (102 pacientes) recibía tratamiento anticoagulante, el 32% (40 pacientes) recibía monodroga (bloqueantes cálcicos, sildenafil, antagonistas de receptores de endotelina 1 o iloprost inhalado) y el 68% (85 pacientes) recibía terapia combinada. El 20.8 % (26 pacientes) fue ingresado en lista de espera para trasplante pulmonar de acuerdo a los criterios de la Sociedad Internacional de Trasplante Corazón y Pulmón14, de los cuales, 7 fueron trasplantados en el seguimiento. Cabe mencionar que los pacientes fueron tratados acorde a la evidencia y según las guías internacionales, y se les ofreció, siempre, el mejor tratamiento específico según la accesibilidad a las diferentes drogas. Los bloqueantes cálcicos y los inhibidores de la fosfodiesterasa 5 estuvieron accesibles desde el momento en que se comenzó el registro, al igual que el iloprost inhalado con indicación para uso compasivo, ya que a partir del año 2005 esta droga estuvo aprobada y disponible en el ámbito comercial. A partir del año 2008 se tuvo acceso al bosentán y al treprostinil y, en el año 2010, al ambrisentán.
Análisis por subgrupos
En las Tablas 2 y 3 se detallan las características clínicas y hemodinámicas por subgrupos. Hubo predominio de mujeres en todos los grupos; la mayoría de los grupos se encontraba en CF avanzada al momento del diagnóstico, excepto los pacientes con HAP portopulmonar los cuales se encontraban, en su mayoría, en CF I/II.
Tabla 3. Características de la población según etiologías

CF: clase funcional; FSVD: función sistólica del ventrículo derecho; PC6M: prueba de caminata de 6 minutos; IC: insuficiencia cardíaca.
*Internaciones por síncope, angina o intercurrencia infecciosa.
El deterioro significativo de la función sistólica del ventrículo derecho estuvo presente en la mayoría de los pacientes del grupo con HAPI (64%) y con cardiopatías congénitas (60%). El grupo con HAPI tuvo una prevalencia de síncope o angina, como síntomas asociados, del 41% y del 25%, respectivamente.
De la población general, hubo una minoría que correspondía a pacientes pediátricos (16 pacientes, 12,8% de la población). La edad promedio fue de 12,5 años (DS 4,4 años), un 44% fue mujer (7 pacientes) y la etiología más frecuente fue la cardiopatía congénita (9 pacientes, 56%). Al momento del seguimiento, el 44% (7 pacientes) presentaba una clase funcional avanzada y deterioro severo de la función ventricular derecha. La mediana de metros caminados en la prueba de la marcha de 6 minutos fue de 312 metros (IIC25-75% 222-378 metros), lo que representa el 47% de la caminata teórica para la edad.
Evolución en el seguimiento
En el seguimiento a 39.1 meses (IIC25-75 21.2- 61.5), la supervivencia total sin necesidad de trasplante pulmonar fue de 63.2%; se registraron 39 muertes y 7 trasplantes, y la supervivencia a 12, 24 y 36 meses fue del 94% (IC95% 88-97), 90% (IC95% 83-94) y 83% (IC95% 75-89), respectivamente (Figura 1). Los pacientes pediátricos presentaron una supervivencia libre de trasplante del 62,5%.

Figura 1. Supervivencia de la población general
En el análisis según etiologías, se observó que los pacientes con cardiopatías congénitas fueron los que presentaron la mayor supervivencia: 95% (IC95% 78-99), 93% (IC95% 75-98) y 93% (IC95% 75-98), a 12, 24 y 36 meses, respectivamente, seguido de los portadores de HAPI: 89% (IC95% 78- 95%), 89% (IC95% 78-95%) y 78% (IC95% 65-87) a 12, 24 y 36 meses, respectivamente. Los pacientes con HAP asociada a colagenopatías presentaron una supervivencia menor con respecto a las otras: 93% (IC95% 63-99), 81% (IC95% 52-93%) y 74% (IC95% 45-89) a 12, 24 y 36 meses, respectivamente (p = 0.01) (Figura 2).
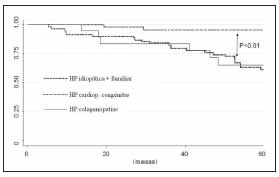
Figura 2. Supervivencia según etiologías. HP: hipertensión pulmonar
En un análisis univariado, las variables basales asociadas a mayor mortalidad fueron: la CF avanzada III/IV (OR 4.7 IC95% 1.9-11.6), una distancia total recorrida <380 m en la PC6M (OR 3.36 IC95% 1.46-7.6), el deterioro significativo de la función sistólica del ventrículo derecho (OR 3.39 IC95% 1.4-7.8), el derrame pericárdico (OR 4.4 IC95% 1.5-12.4), todas las anteriores valor de p < 0,01, y el índice cardíaco (IC) < 2.2 L/min/m2 (OR 2.7 IC 95% 1.1-6.2 p = 0.018) (Tabla 4).
Tabla 4. Variables basales asociadas a mortalidad y/o necesidad de trasplante en el seguimiento. Análisis univariado

CF: clase funcional; IC: índice cardíaco; FSVD: función sistólica del ventrículo derecho; PC6M: prueba de caminata de 6 minutos.
En el análisis multivariado, las exposiciones asociadas a mal pronóstico fueron la CF avanzada III/IV (OR 3.6 IC95% 1.5-8.9, p < 0.01), la distancia total recorrida en la PC6M < 380 m (OR 2.7 IC95% 1.1-6.5 p = 0.023) y la presencia de derrame pericárdico (OR 4.2 IC95% 1.3-14.6 p = 0.021) (Tabla 5).
Tabla 5. Variables basales asociadas a mortalidad y/o necesidad de trasplante en el seguimiento. Análisis multivariado

CF: clase funcional; IC: índice cardíaco; FSVD: función sistólica del ventrículo derecho; PC6M: prueba de caminata de 6 minutos.
Se llevó a cabo un análisis de supervivencia según los grupos de drogas administradas. Los pacientes que recibieron inhibidores de la fosfodiesterasa 5 tuvieron una incidencia acumulada de eventos similar a aquellos que no los recibieron (36% vs 37%, p = 0,8, función de sobrevida long rank p = 0,9); lo mismo ocurrió con el grupo de inhibidores del receptor de endotelina (32% vs 39%, p = 0,46, función de sobrevida long rank p = 0,21). En cambio, los pacientes que recibieron prostanoides tuvieron una incidencia acumulada de eventos mayor que los que no los recibieron (60% vs 25%, p < 0,001, función de sobrevida long rank p = 0,01). Este hallazgo podría estar vinculado a la mayor gravedad clínica de la población bajo tratamiento con prostanoides (deterioro moderado/ severo de la función sistólica del ventrículo derecho: 75% vs 48%, p = 0,005 y bajo índice cardíaco: 53% vs 34%, p = 0,059).
Los casos incidentales tuvieron mayor tasa de eventos en relación a los prevalentes (OR 2.3 IC 95% 1.03-5.1, p = 0.04).
En el seguimiento, los pacientes con CF avanzada (RR 2.4 vs 1,2, p = 0.005), un IC<2.2 L/min/m2 (RR 2.3 versus 1.1, p = 0.008), una distancia total recorrida en la PC6M < 380 m (RR 2.1 versus 1.1 p = 0.01) y el deterioro significativo de la función ventricular derecha (RR 2.8 versus 1.33, p = 0.002) presentaron mayor número de internaciones por falla cardíaca derecha.
Discusión
El presente trabajo representa el primer registro desarrollado en Argentina sobre casos de HAP diagnosticados por CCD, procedimiento considerado como patrón oro diagnóstico. Esta es la primera serie que reproduce, no sólo en Argentina sino en Latinoamérica, los resultados mostrados en las series publicadas en otras regiones del mundo. A pesar de las diferencias étnicas, culturales, ambientales, socioeconómicas y el diferente acceso a los recursos terapéuticos, los resultados hablan de que la enfermedad se manifiesta de manera similar en cualquier región en que se la analice.
El primer registro fue publicado en el año 1991, incluyó más de 150 pacientes portadores de hipertensión arterial primaria-actual idiopática, y fueron los primeros datos epidemiológicos publicados relacionados con la enfermedad4. Luego de más de 20 años, se han realizado grandes avances; en la clasificación actual se reconocen enfermedades que pueden asociarse a HP y se han descubierto líneas de tratamiento que mejoraron francamente la morbilidad y la mortalidad15. Debido a estos cambios, fue necesario crear nuevos registros que mostraran la situación actual. Así, distintos países han publicado su propia casuística y han aportado datos muy valiosos en el entendimiento de esta rara enfermedad5-7, 16-25.
El presente registro confirma el predominio de mujeres en todas las etiologías de HP analizadas, tal cual lo reportado en las otras series. Además, se refuerza la importancia de considerar el diagnóstico en todos los grupos etarios dado que tanto los pacientes pediátricos como aquellos mayores de 60 años se pueden ver afectados por esta enfermedad. La edad promedio de la población fue más baja que en otros registros, esto probablemente esté vinculado a la inclusión de pacientes pediátricos, otras publicaciones no los han contemplado.
Al momento del diagnóstico, la mayoría de los pacientes presentaban marcadores de enfermedad avanzada. Casi el 60% presentaba una CF marcadamente deteriorada; este porcentaje es algo menor al hallado en otros registros5, 7, 23. Este deterioro de la CF estuvo en consonancia con la PC6M basal, que dejó en evidencia un deterioro de la capacidad funcional, y con el ecocardiograma que mostró que más de la mitad de los pacientes tenía deterioro al menos moderado de la función ventricular derecha.
La población de pacientes con respuesta positiva en la prueba aguda de vasorreactividad pulmonar fue baja y se limitó a los pacientes con HAPI. Estos datos son concordantes con los reportados en las otras series y plantean revalorar la indicación de este estudio en pacientes con HP de otras etiologías distintas de la idiopática10, 17-20, 26.
En nuestro registro, la mayoría de los pacientes se encontraban bajo tratamiento con anticoagulación y sildenafil, un menor porcentaje recibía iloprost, bosentán o teprostrinil. El sildenafil constituye, en Argentina, la droga de primera línea de tratamiento, lo cual contrasta con otros registros en donde los antagonistas del receptor de endotelina son las drogas de primera línea de prescripción7. El epoprostenol, a pesar de ser la única droga que ha demostrado beneficios en términos de supervivencia, no se encuentra disponible en Argentina; en su lugar se dispone de treprostinil subcutáneo o intravenoso.
Dentro de los tratamientos de alta complejidad que pueden ofrecerse a un paciente con HP avanzada, se encuentra el trasplante bipulmonar, cardiopulmonar y la septostomía atrial. En la evolución, 7 pacientes fueron sometidos a trasplante bipulmonar. La septostomía es un recurso disponible en nuestro centro, pero no se ha realizado a la fecha.
En el primer registro publicado en el año 1991, la supervivencia media hallada era de sólo 2,8 años4. Desde entonces, ya han transcurrido 22 años. A pesar de contar con drogas más efectivas que actúan sobre vías patogénicas antes ignoradas y que han logrado mejorar la calidad de vida e incluso el pronóstico de esta enfermedad, a la fecha no contamos con una cura definitiva. La supervivencia luego del trasplante pulmonar se ha estimado en 6,7 años según el último reporte de la Sociedad Internacional de Trasplante de Corazón y Pulmón27.
Distintos estudios publicados mostraron una mejoría significativa en el pronóstico de la HP. Así es como el registro francés reportó una supervivencia al año del 88%, el norteamericano del 91% y el británico del 92,7%, en el mismo periodo de tiempo5-7. En nuestra serie, la supervivencia al año fue similar, con una sobrevida al año del 94%.
Al analizar los factores pronósticos y tal cual lo publicado en otros registros y estudios de intervención, se observó que variables como la CF, el deterioro de la función ventricular derecha, la distancia total recorrida en la PC6M y el índice cardíaco reducido, se asociaron a una peor evolución. En cambio, otras variables de valor pronóstico demostrado, como la presión auricular derecha o la respuesta en la prueba aguda de vasorreactividad pulmonar, no tuvieron peso pronóstico de significancia estadística6, 28-31. Estos resultados se deben, probablemente, al número reducido de pacientes de la muestra. Al igual que el registro francés, la población de pacientes incidentales presentó peor evolución que los casos prevalentes.
Cuando se compararon las curvas de supervivencia según las distintas etiologías, se observó que los pacientes con cardiopatías congénitas presentaron una supervivencia mayor que el grupo de HAPI y asociada a enfermedades del colágeno. Este comportamiento diferente ya ha sido resaltado en otras publicaciones; se postula que los pacientes con cardiopatías congénitas tienen mayor adaptación a la HP por una mejor tolerancia del ventrículo derecho que sostiene el aumento de la poscarga desde la vida intrauterina32, 33.
Las limitaciones del presente registro son: la población analizada corresponde a un único centro y el mismo es de referencia, la imposibilidad de haber contado con marcadores biológicos como los péptidos natriuréticos o las troponinas durante todo el periodo analizado, la falta de disponibilidad de estudios genéticos y el escaso número de pacientes pediátricos analizados que sesgan el análisis de los resultados.
Otra limitación es que, no habiendo registros en América Latina, se efectuó una comparación con el mundo occidental. Futuros registros a llevarse a cabo en América Latina nos permitirán realizar análisis de muestras más representativas de la región.
Conclusiones
La HAP es una enfermedad grave y sin tratamiento curativo. La supervivencia de esta serie es similar a la reportada por los registros internacionales actuales y claramente superior a la histórica. Esto podría estar vinculado al avance en el conocimiento de la enfermedad con un perfeccionamiento de la valoración pronóstica y una mayor disponibilidad de tratamientos con acción sobre las vías patogénicas de la enfermedad.
En esta población, variables analizadas de fácil obtención nos permitieron estratificar subgrupos de mayor riesgo.
Al momento del seguimiento, la mayoría de los pacientes se encontraba con marcadores de enfermedad avanzada; la difusión y enseñanza de esta rara enfermedad al resto de la comunidad médica pueden ayudar a la derivación oportuna a grupos especializados.
Conflictos de interés: Los autores declaran que no presentan conflictos de intereses.
Bibliografía
1. Mc Laughlin W, Presberg KW, Doyle RL, et al. American College of Chest Physicians. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004; 126: 78S-92S. [ Links ]
2. Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43: 5S-12S. [ Links ]
3. Ghofrani HA, Barst RJ, Benza RL, et al. Future Perspectives for the Treatment of Pulmonary Arterial Hypertension. J Am Coll Cardiol 2009; 54: S108-S117. [ Links ]
4. D'Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Ann Intern Med 1991; 115: 343-9. [ Links ]
5. Humbert M, Sitbon O, Chaouat A, le Bertocchi M, Habib G, Gressin V. Pulmonary Arterial Hypertension in France. Results from a National Registry. Am J Respir Crit Care Med 2006; 173: 1024-30. [ Links ]
6. Benza RL, Miller DP, Gomberg-Maitland M, et al. Predicting Survival in Pulmonary Arterial Hypertension. Insights From the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010; 122: 164-72. [ Links ]
7. Ling y, Johnson MK, Kiely DG, et al. Changing Demographics, Epidemiology, and Survival of Incident Pulmonary Arterial Hypertension Results from the Pulmonary Hypertension Registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012; 186: 790-6. [ Links ]
8. Simonneau G, Robbins IM, Beghetti M, et al. Updated Clinical Classification of Pulmonary Hypertension. J Am Coll Cardiol 2009; 54: S43-54. [ Links ]
9. Writing Committee Members, McLaughlin VV, Archer SL, Badesch DB, et al. ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension: A Report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: Developed in Collaboration With the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009; 119; 2250-94. [ Links ]
10. Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105-11. [ Links ]
11. The criteria Committee of the New York Heart Association. Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels. 9th ed. Boston, Mass: Little, Brown & Co, 1994, pp 253-6. [ Links ]
12. Enright PL, Sherrill DL. Reference Equations for the Six- Minute Walk in Healthy Adults. Am J Respir Crit Care Med 1998; 158: 1384-7. [ Links ]
13. Geiger R, Strasak A, Treml B, al. Six-Minute Walk Test in Children and Adolescents. J Pediatr 2007; 150: 395-9. [ Links ]
14. Orens JB, Hopkins J, Estenne M, et al. International Guidelines for the Selection of Lung Transplant Candidates: 2006 Update-A Consensus Report From the Pulmonary Scientific Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2006; 25: 745-55. [ Links ]
15. Tuder RM, Abman SH, Braun T, et al. Development and Pathology of Pulmonary Hypertension. J Am Coll Cardiol 2009; 54: S4-S9. [ Links ]
16. Abenhaim L, Moride Y, Brenot F, et al. Appetite Suppressant Drugs ad the risk of primary pulmonary hypertension. N Engl J Med 1996; 335: 609-16. [ Links ]
17. Rich S, Rubin L, Walker AM, Schneeweiss S, Abenhaim L. Anorexigens and pulmonary hypertension in The Unites States: results for the surveillance of North American Pulmonary Hypertension. Chest 2000; 117; 870-4. [ Links ]
18. Appelbaum L, Yigla M, Bendayan D, et al. Primary Pulmonary Hypertension In Israel: A National Survey. Chest 2001; 119; 1801-6. [ Links ]
19. Stricker H, Domenighetti G, Popov W, et al. Severe pulmonary hypertension: data from the Swiss Registry. Swiss Med WKLY 2001; 131: 346-50. [ Links ]
20. Walker AM, Langleben D, Korelitz JJ, et al. Temporal trends and drug exposures in pulmonary hypertension: An American experience. Am Heart J 2006; 152: 521-6. [ Links ]
21. Peacock AJ, Murphy NF, McMurray JJV, Caballero L, Stewart S . An Epidemiological study of Pulmonary Arterial Hypertension. Eur Respir J 2007; 30: 104-9. [ Links ]
22. Jing ZCH, Xu Xq, Han XI, et al. Registry and Survival Study in Chinese Patients With Idiopathic and Familial Pulmonary Arterial Hypertension. Chest 2007; 132; 373-9. [ Links ]
23. Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary Arterial Hypertension Baseline Characteristics From the REVEAL Registry. Chest 2010; 137 (2): 376-87. [ Links ]
24. Zhang R, Dai LZ, Xie WP, et al. Survival of Chinese Patients with Pulmonary Arterial Hypertension in the Modern Management Era. Chest 2011; 140: 301-9. [ Links ]
25. Hurdman J, Condliffe R, Elliot CA, et al. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur Respir J 2012; 39: 945-55. [ Links ]
26. Klings ES, Hill NS, Ieong MH, Simms RH, Korn JH, Farber HW. Systemic sclerosis-associated pulmonary hypertension: short- and long-term effects of epoprostenol (prostacyclin). Arthritis Rheum 1999; 42: 2638-45. [ Links ]
27. Hertz MI. The Registry of the International Society for Heart and Lung Transplantation-Introduction to the 2012 Annual Reports: New leadership, Same Vision. J Heart Lung Transplant 2012; 31: 1045-95. [ Links ]
28. Sitbon O, Humbert M, Nunes H, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002; 40: 780-8. [ Links ]
29. Humbert M, Sitbon O, Chaouat A, et al. Survival in Patients With Idiopathic, Familial, and Anorexigen-Associated Pulmonary Arterial Hypertension in the Modern Management Era. Circulation 2010; 122: 156-63. [ Links ]
30. Eysmann SB, Palevsky HI, Reichek N, Hackney K, Douglas PS. Two-dimensional and Doppler-echocardiographic and cardiac catheterization correlates of survival in primary pulmonary hypertension. Circulation 1989; 80: 353-60. [ Links ]
31. Tei C, Dujardin KS, Hodge DO, et al. Doppler echocardiographic index for assessment of global right ventricular function. J Am Soc Echocardiogr 1996; 9: 838-47. [ Links ]
32. Beghetti M, Galiè N. Eisenmenger Syndrome. A Clinical Perspective in a New Therapeutic Era of Pulmonary Arterial Hypertension. J of Am Coll of Cardiol 2009; 53: 733-40. [ Links ]
33. Diller GP, Gatzoulis MA. Pulmonary. Vascular Disease in Adults with Congenital Heart Disease. Circulation 2007; 115; 1039-50. [ Links ]

