










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. v.4 n.1 Ciudad Autónoma de Buenos Aires ene./mar. 2009
SIMPOSIO INTERNACIONAL DE HIPERTENSIÓN PULMONAR
Mecanismos fisiopatológicos involucrados y diagnóstico de la hipertensión arterial pulmonar
Juan Antonio Mazzei*
* Profesor de Medicina. Facultad de Medicina. Universidad de Buenos Aires (UBA). Ciudad de Buenos Aires. República Argentina.
Regente del American College of Chest Physicians. Delegado Nacional de la European Respiratory Society.
Director de la Escuela Argentina de Medicina Respiratoria. Presidente de la Fundación Argentina del Tórax.
Correspondencia: Dr. Juan Antonio Mazzei.
Hospital de Clínica José de San Martín. Av. Córdoba 2351 - CP: 1120 - Ciudad Autónoma de Buenos Aires. República Argentina.
E-mail: jamazzei@fibertel.com.ar
Recibido: 10/12/2008
Aceptado: 10/03/2009
Palabras clave: Hipertensión arterial pulmonar; Fisiopatología; Endotelina; Oxido nítrico; Prostaciclina; Mutaciones genéticas
Los mecanismos fisiopatológicos de la hipertensión arterial pulmonar (HAP) abarcan una serie de modificaciones vasculares que producen un aumento de la resistencia vascular pulmonar (RVP).
Las modificaciones vasculares que se producen en la hipertensión arterial pulmonar incluyen: la vasoconstricción, la proliferación del músculo liso, la inflamación, la apoptosis endotelial, la proliferación endotelial resistente a la apoptosis, la fibrosis, la trombosis in-situ, y finalmente, las lesiones plexiformes (Figura 1). Todas estas modificaciones producen una alteración de la RVP, que podemos explicar aplicando la ley de Ohm:
ΔP = QP x RVP
ΔP = Pap - Pvp
Pap - Pvp = QP x RVP
Pap = (QP x RVP) + Pvp
PA media = (QP x RVP) + Pcp
Donde la diferencia de presión (ΔP) es directamente proporcional a la RVP si el flujo vascular pulmonar (QP) es constante (volumen minuto pulmonar = VMP). Esta diferencia de presión es la que se establece entre la presión arterial pulmonar (Pap) y la presión venosa pulmonar (Pvp). Por lo tanto, la diferencia de presión es el producto del flujo pulmonar (QP) por la RVP. Entonces, la presión arterial media va a ser proporcional al flujo por la RVP más la presión capilar pulmonar o wedge (Pcp). A medida que aumenta la RVP, se presentan las distintas fases de esta enfermedad, clasificándola en diferentes etapas: la etapa pre-clínica, la etapa sintomática/estable y la etapa progresiva/declinante1.
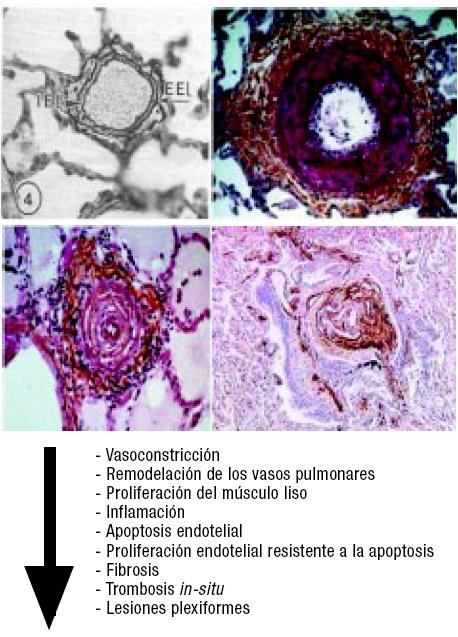
Figura 1. Modificaciones vasculares de la hipertensión arterial pulmonar.
Como vemos en la Figura 2, el volumen minuto en ejercicio disminuye progresivamente a medida que avanza la enfermedad, mientras que el volumen minuto en reposo se mantiene hasta la etapa declinante de la misma. La presión arterial pulmonar aumenta en la fase pre-clínica y se mantiene elevada en el resto de la evolución.
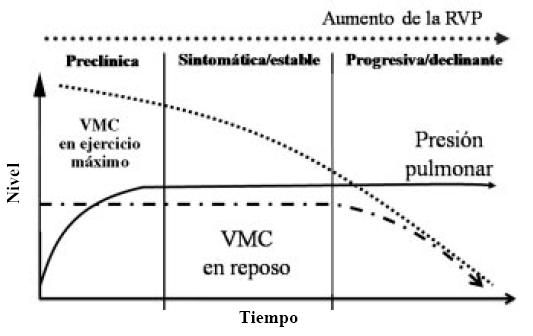
Figura 2. Historia natural de la hipertensión arterial pulmonar. VMC: Volumen minuto cardíaco. RVP: Resistencia vascular pulmonar1.
En la investigación de los mecanismos fisiopatológicos de la HAP, hay dos hitos muy importantes. El primero es el descubrimiento de mutaciones genéticas específicas; y el segundo, el reconocimiento de componentes vasoactivos y factores de crecimiento de origen principalmente endotelial que aumentan o disminuyen el riesgo (Tabla 1).

La primera base genética que se descubrió es la mutación heterocigota del gen del receptor tipo II de la proteína ósea morfogenética (BMP), que se ubica en el cromosoma 2 en la porción q33 y que codifica para el receptor tipo II: un miembro de la familia del factor transformador del crecimiento β (TGF-β) (Figura 3).

Figura 3. Proteínas óseas morfogenéticas. ~20 miembros de la familia. Forma 30-38 kDa dímeros. Estructura nudos de cisteína.
La proteína ósea morfogenética es una sustancia sumamente importante que participa en distintas funciones; como por ejemplo, la formación de hueso y cartílago, la formación embrionaria mesodérmica, la definición del patrón dorso-ventral, la organogénesis sobre todo del riñón y de los pulmones, la neurogénesis, la angiogénesis y la diferenciación vascular. Y que posee un receptor que puede mutar2.
Cuando se produce una mutación del receptor, si bien no es la única mutación que se ha descubierto, se desencadenan modificaciones.
Pocos años después del descubrimiento de la primera mutación, se encontró una segunda que induce el desarrollo de la HAP en el síndrome de telangiectasia hemorrágica hereditaria (Figura 4).
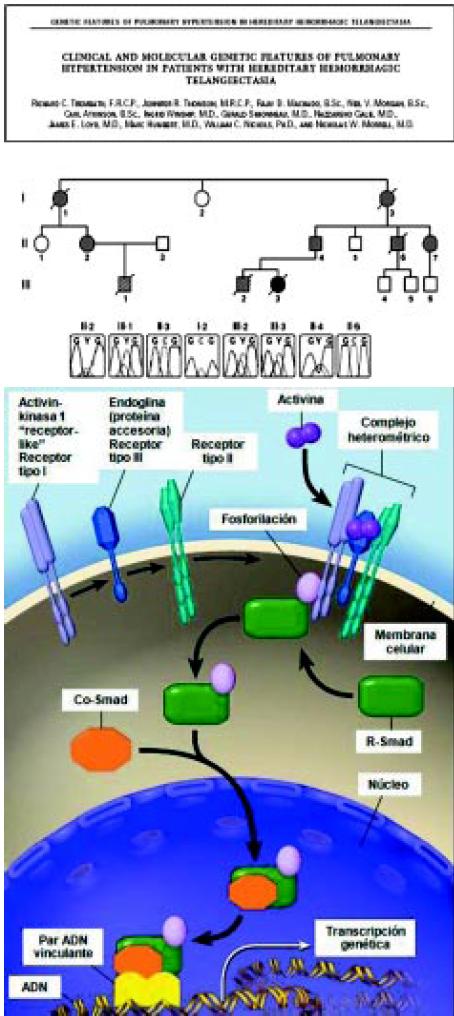
Figura 4. Segregación de la mutación de citosina a timina en la posición 1450 en ALK1 con hipertensión pulmonar y telangiectasia. Hemorrágica hereditaria en la familia 12.
El receptor de la proteína morfogenética (BMPR) posee dos partes: el BMPR I y el BMPR II. El segundo es el que se halla alterado. Cuando se produce la activación del receptor, el BMPR I activa al BMPR II que a su vez fosforila sustancias, los smad. Estas sustancias fosforiladas se unen a otras sustancias, entrando a nivel del núcleo y uniéndose al ADN, produciendo una transcripción genética. Al producirse la mutación del receptor, la función normal de la angiogénesis se altera (Figura 5)3.
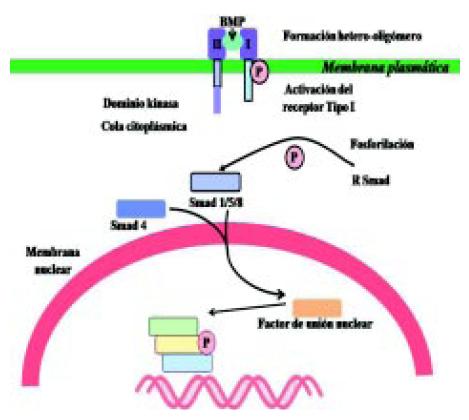
Figura 5. Transcripción genética del receptor de la proteína morfogenética (BMP) en la hipertensión arterial pulmonar3.
En la telangiectasia hemorrágica hereditaria, existe una alteración del receptor ALK-1, produciéndose una alteración de la relación de distintos tipos de ligandos receptores reguladores de smad, produciéndose una alteración de la angiogénesis (Figura 6)4.

Figura 6. Visión actual simplificada del rol del receptor de la proteína-II (BMPR-II) en la señalización del FCT-β4.
Se acepta actualmente que la mutación del BMPR-II es una condición necesaria, pero no suficiente para el desarrollo de la HAP. La enfermedad genética tiene una penetración menor del 50%, y sólo entre el 10% y el 20% de los que tienen la mutación desarrollan HAP. Hay ejemplos de mellizos idénticos con mutaciones del BMPR-II, donde sólo uno desarrolla HAP.
Sería importante explicar cómo actúan otros desencadenantes conocidos para la hipertensión arterial severa, como la dexfenfluramina y aceites tóxicos, y existen otros desencadenantes adicionales que puedan ser genéticos o ambientales.
¿Cuál es entonces la patogenia más aceptada actualmente?
Existe una modificación del receptor II de la proteína morfogenética ósea (mutación genética del BMPR-II), y ello predispone a la vasoconstricción y remodelación vascular. Algunos genes pueden ampliar esta predisposición, tal es el caso de los genes de la endotelina 1 (ET 1), de la serotonina y de los transportadores de la serotonina, el tromboxano, o del factor de crecimiento derivado de las plaquetas, o algunas noxas como los anorexígenos, haciendo que esta mutación genética se exprese. Por otro lado, pueden existir otros genes como el de las prostaciclina, la sintetasa de óxido nítrico endotelial, el factor natriurético atrial, la adrenomedulina, o el factor de crecimiento vascular derivado del endotelio, que tienen una acción contraria a la anterior. De la interacción de esto, se podrá desarrollar la HAP (Figura 7)3.

Figura 7. Interacciones que podrían desarrollar la hipertensión arterial pulmonar (HAP)3.
Es decir que la mutación del BMPR II es el primer golpe, pero tiene que haber un segundo golpe, a partir de la activación de los otros genes para que se pueda producir la remodelación vascular y una vasculatura inestable, con el desarrollo de las lesiones anatomopatológicas que vimos al comienzo de este compendio.
Es decir, hay un balance entre la vasoconstricción y la vasodilatación. La vasoconstricción, que a su vez lleva a la proliferación endotelial y de músculo liso, está mediada por: la ET 1, la angiotensina II, la serotonina, el tromboxano, el factor de crecimiento derivado de las plaquetas. Y la vasodilatación y la anti-proliferación están inducidas por: la prostaciclina, el óxido nítrico, el factor natriurético atrial, el péptido intestinal vasoactivo (VIP) y el polipéptido activador de la adenilil ciclasa de la pituitaria (PACAP), la adrenomodulina y el factor de crecimiento vascular endotelial (Figura 8).

Figura 8. Balance endotelial: vasoconstricción - vasodilatación.
Es decir, cuando predomina la vasoconstricción, habrá una disminución del flujo, y una proliferación vascular (Figura 9). Por el contrario, cuando predomina la vasodilatación, habrá un aumento de flujo y una anti-proliferación vascular (Figura 10).

Figura 9. Balance endotelial: vasoconstricción.

Figura 10. Balance endotelial: vasodilatación.
El sistema endotelina es un sistema sumamente importante, que se ha estudiado en los últimos años, y que está generado por la BIG-endotelina o pro-endotelina, sustancia producida por la célula del endotelio vascular. Esta es una sustancia inactiva, que es transformada por la enzima convertidora de la endotelina en ET 1. La ET 1 tiene distintas acciones. Por un lado, a nivel de la célula endotelial, activa el receptor de la endotelina B (ETB) que produce sustancias como el óxido nítrico (NO) y la prostaciclina (inhibidores de la producción de endotelina). Pero por otro lado, la ET 1, actuando sobre los receptores de la célula muscular lisa (tanto el A como el B), produce vasoconstricción y proliferación celular (Figura 11)5. La ET 1 cumple un papel crítico en las células vasculares pulmonares de la HAP severa (Figura 12)6.
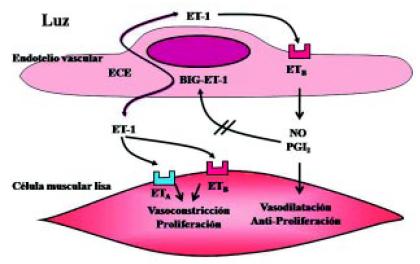
Figura 11. Sistema endotelina en el tejido vascular de la hipertensión arterial pulmonar (HAP). ET-1: Endotelina 1. BIG-ET-1: Proendotelina 1. ECE: Enzima convertidora de la endotelina. NO: óxido nítrico. PGI2: prostaciclina I2. Adaptado de Dupuis5.

Figura 12. Expresión aumentada de la ET-1 vascular pulmonar en la hipertensión pulmonar severa6.
En la actualidad, existen tres caminos terapéuticos, en base a estos conocimientos de los mecanismos fisiopatológicos, si bien existen otros que están siendo estudiados (Figura 13).
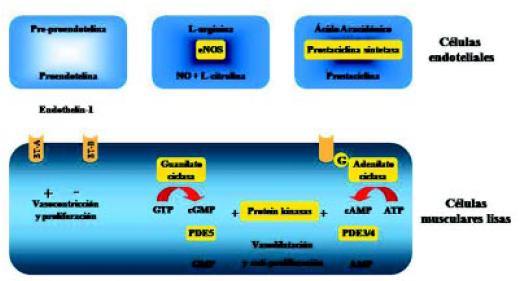
Figura 13. Caminos principales involucrados en el control del tono y remodelado vascular.
El primero de estos mecanismos es el de la endotelina: la pro-endotelina o BIG-endotelina transformada en ET 1 actúa uniéndose a los receptores de endotelina A y B. El receptor A es estimulante, produciendo vasoconstricción y proliferación, y el receptor B es inhibidor. Este mecanismo se encuentra aumentado en la HAP. El segundo mecanismo es el de la enzima óxido nítrico-sintetasa, que a partir de la arginina, produce NO, el cual por el mecanismo de la guanidil-ciclasa produce vasodilatación y anti-proliferación. La producción de NO está disminuida en la HAP.
El tercer mecanismo es la disminución de la producción de prostaciclina por las células endoteliales. Los niveles bajos de prostaciclina producen vasoconstricción y proliferación vascular. En un clásico trabajo de Humbert, se muestran estos tres caminos patogénicos y terapéuticos: a) el camino de la endotelina con aumento de la producción de la misma; b) el camino del NO con disminución de su concentración celular, y c) el camino de la prostaciclina con disminución de su producción (Figura 14)7.
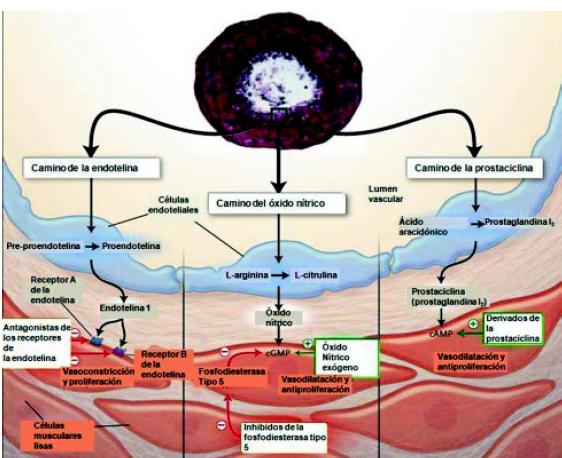
Figura 14. Caminos alterados en la hipertensión arterial pulmonar7.
Entre otros caminos patogénicos y terapéuticos en estudio, existe el camino serotoninérgico, donde hay algunos nuevos medicamentos que se están utilizando, en el cual a partir de la PO2 disminuida (es decir la hipoxia) o a partir de anorexígenos, se puede producir un aumento del transporte de serotonina con proliferación celular y remodelamiento vascular (Figura 15)8.
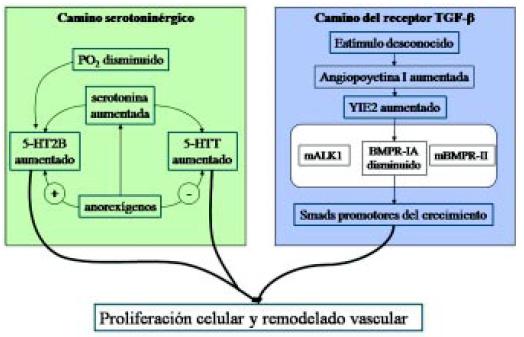
Figura 15. Camino serotoninérgico y camino del receptor TGF-β en la producción de la hipertensión arterial pulmonar8.
En conclusión, para el desarrollo de HAP debe haber una predisposición genética debida a algunas mutaciones, o puede haber algún tipo de interacción ambiental (como anorexígenos, aceites tóxicos, infección por el virus de inmunodeficiencia humana -VIH-), y debe haber algún otro tipo de noxas o de expresión de genes, como el aumento de la serotonina o del transporte de serotonina, la disminución del NO, de la prostaciclina, el aumento de la endotelina o del tromboxano, que a su vez se traducen en un aumento de la producción de elastasa, de metaloproteinasa de la matriz, produciendo un aumento del colágeno y de la elastina. Ultimamente, se han encontrado cambios inflamatorios a nivel de las células y del tejido de las arterias pulmonares que podrían contribuir a la patogenia (Figura 16)9.

Figura 16. Cambios inflamatorios a nivel de las células y del tejido de las arterias pulmonares que podrían contribuir a la hipertensión arterial pulmonar9.
1. Braunwald E, Fauci AS, Isselbacher KJ, Martin JB, Wilson JD, Kasper DL, Hauser SL. Harrison's Principles of Internal Medicine. 14th ed. México: McGraw Hill Interamericana, 1998. [ Links ]
2. Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, Simonneau G, Galie N, Loyd JE, Humbert M, Nichols WC, Berg J, Manes A, McGaughran J, Pauciulo M, Wheeler L, Morrell NW. Clinical and Molecular Genetic Features of Pulmonary Hypertension in Patients with Hereditary Hemorrhagic Telangiectasia. N Engl J Med 2001;345:325-334. [ Links ]
3. Said SI. Mediators and modulators of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 2006;291:547-558. [ Links ]
4. Wilkins MR, Gibbs JS, Shovlin CL. A gene for primary pulmonary hypertension. The Lancet 2000;356:1207-1208. [ Links ]
5. Dupuis J. Endothelin-receptor antagonists in pulmonary hypertension. The Lancet 2001;358:1113-1114. [ Links ]
6. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib, H et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993;328:1732-1739. [ Links ]
7. Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004;351:1425-1436. [ Links ]
8. Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson JL, Loscalzo JL. Harrison's Principles of Internal Medicine. 17th ed. México: McGraw Hill Interamericana, 2008. [ Links ]
9. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O, Voelkel NF, Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43:13S-24S. [ Links ]

