










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. v.4 n.1 Ciudad Autónoma de Buenos Aires ene./mar. 2009
SIMPOSIO INTERNACIONAL DE HIPERTENSIÓN PULMONAR
Bosentan
Rol en el tratamiento de la hipertensión arterial pulmonar
Juan Carlos Grignola*, Miguel Angel Gómez Sánchez**
* Departamento de Fisiopatología. Facultad de Medicina. Universidad de la República. Montevideo. República Oriental del Uruguay.
** Unidad de Insuficiencia Cardíaca e Hipertensión Pulmonar. Servicio de Cardiología. Hospital Universitario Doce de Octubre. Madrid. España.
Correspondencia: Dr. Miguel Angel Gómez Sánchez.
Unidad de Insuficiencia Cardíaca e Hipertensión Pulmonar. Servicio de Cardiología, Hospital Universitario Doce de Octubre.
Ctra. Andalucía Km 5,400 (28041). Madrid-España.
Teléfono: (0034) 913 908289
Fax: (0034) 913 908669
E-mail: mangomesz@telefonica.net
Recibido: 10/12/2008
Aceptado: 10/03/2009
Palabras clave: Hipertensión arterial pulmonar; Bosentan; Disfunción ventricular derecha
Introducción
Importancia de la hipertensión pulmonar
Las alteraciones cardiovasculares más frecuentes son: la enfermedad coronaria, la hipertensión arterial sistémica y la hipertensión pulmonar (HP).
La HP es una complicación habitual en el paciente con cardiopatía o valvulopatía izquierda. Cerca de un 40% de pacientes con enfermedad del lado izquierdo del corazón presenta HP y es la causa más frecuente de la misma (50%). Es un marcador pronóstico y tiene importantes implicaciones terapéuticas. En la enfermedad pulmonar obstructiva crónica (EPOC), la HP también es una complicación frecuente (cerca del 15-20% de las HP), asociándose su presencia a menor sobrevida y peor evolución clínica. En ambas situaciones, el pronóstico de la enfermedad subyacente depende del grado de HP acompañante. La hipertensión arterial pulmonar (HAP), entidad menos frecuente, bien en su forma idiopática, heredable o asociada a situaciones clínicas diversas como las conectivopatías, cardiopatías congénitas, infección por virus de inmunodeficiencia humano (VIH) entre otras, puede afectar a 16 personas por millón de habitantes. Un cuarto grupo de HP debida a enfermedad trombótica crónica (5-10%) y finalmente la HP no aclarada o de mecanismos desconocidos1.
En los últimos años, se han producido importantes avances en el diagnóstico y tratamiento de la HP que han logrado una mejoría significativa en la supervivencia de esta enfermedad. Estos avances han sido impulsados por 4 reuniones mundiales de expertos. Las 2 primeras promovidas por la Organización Mundial de la Salud (OMS), tuvieron lugar en Ginebra (1975)2, donde se elaboró la primera clasificación de la enfermedad vascular pulmonar, diferenciando arteriopatía pulmonar de la hipertensión venosa pulmonar y enfermedad tromboembólica, estableciéndose el primer registro que permitió conocer la historia natural de la enfermedad3, y en Evian (Francia) en 19984, que introdujo una clasificación fisiopatológica de la enfermedad con consecuencias importantes sobre el tratamiento de estos pacientes. Posteriormente, y en base al importante avance en el conocimiento de la genética y biopatología de la HP, siguieron las reuniones de Venecia en 2003, que consolidó con algunos cambios la clasificación anterior (se abandonó el término de HP primaria, se acuñó un subgrupo denominado HP familiar, se reclasificó la enfermedad venooclusiva pulmonar y la hemangiomatosis capilar pulmonar, se actualizaron los factores de riesgo y condiciones asociadas a la HP y se revisó la clasificación de la cardiopatías congénitas asociadas a shunt sistémico-pulmonar) y dio las primeras directrices sobre el empleo de los fármacos vasodilatadores pulmonares selectivos empleados con éxito hasta ese momento5, y finalmente la de Dana Point (California, EE.UU.) celebrada en febrero de 2008, donde se establecieron bases más racionales para la definición de HP.
Definición y clasificación de la hipertensión pulmonar
La HP se define por el incremento anómalo de la presión en la arteria pulmonar. En la reunión de Venecia (2003), se acordó por consenso que la HP estaba definida por una presión media de la arteria pulmonar (PMAP) mayor a 25 mm Hg en reposo o mayor de 30 mm Hg en esfuerzo. En base a los datos hemodinámicos de 996 adultos sanos recopilados de más de 30 publicaciones cuidadosamente seleccionadas, Olchewsky y col. (datos no publicados) establecieron unas bases más racionales para la definición de la HP, teniendo en cuenta que el valor medio de la PMAP en reposo fue de 14 ± 3,3 mm Hg (Tabla 1). Destacándose que ni la edad ni el sexo parecen influir significativamente en estas cifras. A diferencia de lo que ocurre con la PMAP en reposo, la edad sí parece influir en la PMAP con el esfuerzo, así como el grado de esfuerzo (Tabla 2).


El esquema general de la reunión de Venecia (2003) se mantuvo en la clasificación de la reciente reunión de Dana Point6 (2008), aunque con algunos cambios:
• En el grupo I de HAP, se mantiene el término de HAP idiopática para los casos esporádicos sin causa conocida. Se ha cambiado el término “familiar” por el más ajustado de “heredable”, pues dada la posibilidad de mutaciones espontáneas de novo y la variabilidad en la penetrancia de las alteraciones genéticas, es posible la presencia de este tipo de HAP asociadas a genopatías sin que se dé una agregación familiar. Por otro lado, un cierto número de entidades poco frecuentes incluidas en este grupo en la clasificación anterior (HP asociada a trastornos tiroideos, glucogenosis, enfermedad de Gaucher, telangiectasia hemorrágica hereditaria, hemoglobinopatías, síndromes mieloproliferativos, esplenectomía) se ha trasladado al grupo V de miscelánea, ante la ausencia de pruebas del componente “arterial” en las mismas. Lo que a la vez retiró la indicación de tratamiento vasodilatador pulmonar en estas entidades. Lo contrario ha sucedido con la esquistosomiasis, que ha pasado a pertenecer a este grupo ante la evidencia de una fisiopatología y respuesta a fármacos similar a otras formas de HAP, a pesar de originarse en una parasitosis. Dos entidades muy particulares por su fisiopatología, mala respuesta terapéutica y rápida evolución son la enfermedad venooclusiva y la hemangiomatosis capilar. Puesto que sus características no reunían satisfactoriamente las de los grupos I y II, se les creó un grupo diferente, el grupo I.
• En el grupo II, HP asociada a insuficiencia cardíaca, se ha introducido una referencia explícita al tipo de disfunción ventricular izquierda. De especial importancia es la HP asociada a disfunción diastólica, muy frecuente en la clínica (en general con grados leves-moderados de HP), que a menudo causa problemas de diagnóstico diferencial con la HAP, cuyo enfoque terapéutico es muy diferente. Para llegar a diferenciar las 2 entidades, además de considerar los factores de riesgo para disfunción diastólica, es necesario realizar un cateterismo derecho, en el que una presión capilar pulmonar superior a 15 mm Hg será indicativa del origen cardiogénico de la HP.
• En el grupo III, HP asociada a neumopatías, no se han introducido cambios significativos.
• En el grupo IV, HP tromboembólica, se ha eliminado la diferenciación un tanto artificial referente a la localización de los trombos (proximales, susceptibles de endarterectomía quirúrgica, frente a los distales, susceptibles de tratamiento con vasodilatadores pulmonares selectivos). Parece claro que se trata de una sola entidad fisiopatológica y cada vez es más frecuente que estos pacientes reciban un tratamiento mixto en el que los vasodilatadores pueden administrarse antes, durante y/o después de la endarterectomía.
• En el grupo V, que recoge una miscelánea de entidades cuya relación con la HP es poco clara o debida a múltiples causas, se ha visto engrosado por una lista de entidades que figuraban en el grupo I de la clasificación anterior, como se señaló precedentemente.
Fisiopatología de la hipertensión pulmonar: rol del sistema de la endotelina
La HP es una enfermedad crónica caracterizada por la remodelación progresiva vascular y la oclusión de las arteriolas pulmonares, que conlleva a la disfunción del ventrículo derecho (VD) y muerte por fracaso del mismo. La remodelación de la vasculatura pulmonar incluye proliferación intimal, hipertrofia de la capa media, fibrosis adventicial, necrosis fibrinoide y desarrollo de las lesiones plexiformes patognomónicas. La comprensión de la patogenia de la HAP ha progresado considerablemente en los últimos años7. Más allá del tipo de injuria o insulto vascular inicial, el resultado final es la disfunción de la célula muscular vascular lisa y la disfunción de la célula endotelial. Esta última resulta de un disbalance de las moléculas que controlan la homeostasis vascular y mantienen el tono vascular. En general, los niveles de las moléculas pro-vasodilatadoras y antiproliferativas, como el óxido nítrico (ON) y la prostaciclina, están disminuidas; mientras que las moléculas pro-vasoconstrictoras y pro-proliferativas, como el tromboxano A2 y la endotelina-1 (ET-1) están aumentadas. Ello ha permitido desarrollar estrategias terapéuticas cuyo objetivo es restablecer el balance de ambos grupos de moléculas (Figura 1). En este artículo nos centraremos en el rol del sistema de la ET-1 y el bloqueo del mismo mediante un antagonista no selectivo de los receptores de la ET-1, bosentan.
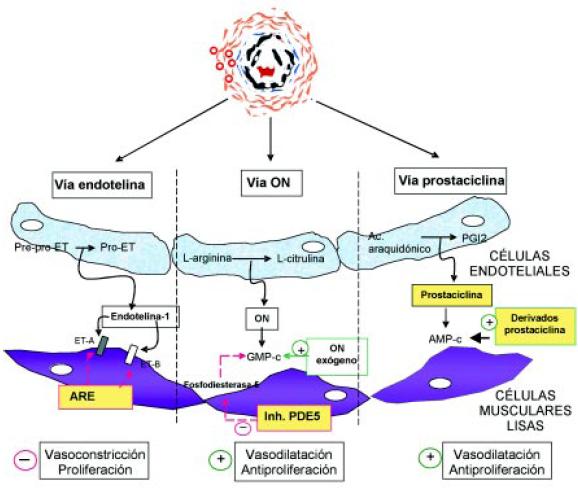
Figura 1. Objetivos terapéuticos en la hipertensión arterial pulmonar (HAP). Arriba: sección transversal de una arteria pulmonar pequeña (< 500 µm) de un paciente con HAP severa. AMP-c: adenosina monofosfato cíclico. GMP-c: guanosina monofosfato cíclico. ET: endotelina. ETA y ETB: receptores A y B de la ET. PDE5: fosfodiesterasa tipo 5. PGI2: prostaglandina I2.
Biología del sistema de la endotelina
Desde su descubrimiento en 1988, la ET-1 ha sido implicada en forma creciente en la patogenia de la HAP8. La ET-1 está constituida por una cadena de 21 aminoácidos. Es producida fundamentalmente por el endotelio vascular, y en menor medida por otros tipos celulares incluyendo las células musculares lisas de la arteria pulmonar y los fibroblastos pulmonares9. Existen varios promotores que pueden estimular la síntesis de la ET-1, incluyendo la hipoxia, factores de crecimiento, citoquinas, estrés de cizallamiento, trombina y angiotensina II. Curiosamente su síntesis es inhibida por el ON y la prostaciclina, 2 moléculas cuya menor concentración contribuye en la fisiopatología de la HAP. Estudios pre-clínicos sugieren que los pulmones son el principal sitio de producción de la ET-1. Los pulmones no sólo producen ET-1, sino que también la quitan de la circulación, con un 47% de extracción en el primer paso a través éstos. En sujetos normales, no existe un gradiente arteriovenoso de ET-1 significativo a través del pulmón, ya que el aclaramiento y la secreción en el plasma están balanceados9. Existe clara evidencia de la activación del sistema de la ET en varios modelos pre-clínicos de HP, así como también en todas las categorías de HP en el ser humano. Tanto la ET-1 plasmática como la expresión en los vasos pulmonares están aumentadas en varios tipos de HP (idiopática, HP asociada a enfermedad de tejido conectivo, cardiopatía congénita e infección por VIH). El nivel de ET-1 se correlaciona con la severidad clínica (test de 6 minutos y sobrevida) y hemodinámica (resistencia vascular periférica y PAPM) de la enfermedad8,10,11 (Figuras 2 y 3). Sin embargo, su significado fisiopatológico no está aclarado. Los niveles de ET-1 parecen ser un marcador de la severidad de la disfunción endotelial más que su desencadenante; así la perfusión crónica de ET-1 en ratas hasta alcanzar los niveles encontrados en pacientes con HP reduce la vasorreactividad vascular pulmonar, pero no es capaz de inducir la HP12. Más aún, la sobreexpresión de ET-1 en animales transgénicos tampoco resulta en HP13. Los efectos biológicos de la ET-1 se deben a su unión con dos tipos de receptores de endotelina A y B (ETA y ETB). Ambos receptores se expresan en la capa media de las arterias pulmonares a nivel de las células musculares lisas y fibroblastos, donde median vasoconstricción, proliferación y fibrosis. El ETB se encuentra fundamentalmente en el endotelio y su estimulación provoca liberación de ON, determinando una moderada vasodilatación transitoria, además de ser el encargado del aclaramiento plasmático de la ET-1 e inhibir la enzima convertidora de ET-1 (requerida para producir ET-1 madura) (Figura 4). La densidad de receptores en las arterias distales y el parénquima pulmonar es 2 veces mayor (p < 0,05) en pacientes con HP con respecto a sujetos control, a la vez que si bien predominan los ETA, la relación ETA/ETB disminuye significativamente, debido a un aumento porcentual de los ETB hacia los vasos distales (36 ± 3 vs 3 ± 1%)9. Otro concepto importante a tener en cuenta es que la distribución relativa de los receptores ETA y ETB pueden depender del tipo de HP. Bauer y col. demostraron que, en la HP trombótica crónica, la expresión de los ETB estaba aumentada; mientras que la de los ETA no se modificó14. A su vez, debemos conocer cual fracción del ETB está alterada, la endotelial (aclaramiento de ET-1 y vasodilatación) o la muscular o fibroblástica, ya que la estimulación de esta última sub-población conlleva a la vasoconstricción, proliferación y fibrosis en pacientes con HP. Clásicamente, se ha sugerido que el endotelio podría ser disfuncionante, resultando en la pérdida de función o disminución de la expresión del ETB endotelial. Recientemente, Langleben y col. demostraron que el aclaramiento de la ET-1 por los ETB estaba modestamente reducido en pacientes con HAP idiopática, asociada a enfermedad del tejido conectivo y en la HP trombótica crónica, sugiriendo que los altos niveles de ET-1 observados en la HAP se deban predominantemente a un exceso de síntesis más que a una caída de su aclaramiento15.

Figura 2. Correlación de los niveles plasmáticos de ET-1 y la severidad hemodinámica de la hipertensión pulmonar. (Modificado de Rubens y col., 200110).

Figura 3. Correlación entre los niveles plasmáticos de ET-1 y la sobrevida en la hipertensión arterial pulmonar idiopática (Adaptado de Galié y col., 199611). IC: índice cardíaco. RVP: resistencia vascular pulmonar.
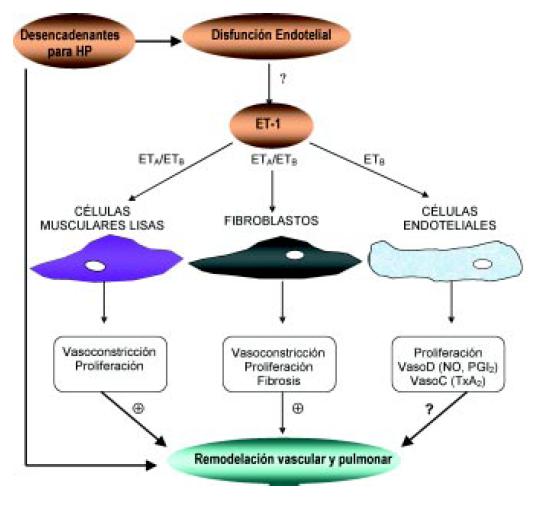
Figura 4. Efectos de la ET-1 en la hipertensión arterial pulmonar. (Modificado de Dupuis & Hoeper, 20089).
Bosentan: antagonista dual o no selectivo de los receptores de la endotelina
Bosentan es un antagonista competitivo no selectivo de los receptores de endotelina (Figura 5). Previene la proliferación celular mediada por la ET-1 en tejido humano ex-vivo. Este efecto antiproliferativo es significativamente mayor en las células musculares lisas de los vasos pulmonares de pacientes con HAP, que en el mismo tipo de células procedentes de sujetos sanos.

Figura 5. Estructura química del bosentan.
La máxima concentración plasmática y el área debajo de la curva luego de dosis simples o múltiples v/o es proporcional a la dosis utilizada dentro del rango de dosificación (≤ 500 mg/día)15. La viabilidad absoluta es del 50% luego de la administración v/o. Presenta una baja tasa de absorción debido a su escasa hidrosolubilidad (tiempo a la concentración máxima 3 hs). Se une fundamentalmente a la albúmina hasta un 98% y luego de 3 a 5 días de ser administrado alcanza niveles de los parámetros farmacocinéticos estables. Se metaboliza en el hígado mediante la vía de la citocromo P450 (CYP450): la CYP2C9 y la CYP3A4. Primariamente sufre hidroxilación y demetilación dando lugar a los metabolitos Ro 48-5033(capaz de producir hasta el 20% de los efectos de bosentan) y Ro 47-8634 (inactivo) para luego de una posterior metabolización formar el metabolito Ro 64-1056 inactivo, que se elimina por las heces. Luego de la administración de múltiples dosis se produce la auto-inducción de CYP2C9 y CYP3A4, alcanzo el máximo a los 4-5 días.
Debido a que es sustrato y a la vez inductor de las isoenzimas hepáticas CYP2C9 y CYP3A4, bosentan puede interactuar con otros sustratos de la CYP2C9 (S-warfarina, glibenclamida, anticonceptivos orales) e inductores de la CYP2C9 o 3A4 (carbamacepina, glibenclamida) o inhibidores (ketoconazol, itraconazol, fluconazol, ritonavir, voriconazol, eritromicina, ciclosporina)15. El uso de bosentan está contraindicado en pacientes que reciban glibenclamida, ciclosporina o anticonceptivos orales en forma concomitante. Otras drogas con las que puede interactuar son el tracolimus y sirolimus, así como la rifampicina.
Evidencia de la eficacia clínica de bosentan en la hipertensión pulmonar
Fue el primer tratamiento oral disponible para pacientes con HAP. Fue aprobado por la Food and Drug Administration (FDA) en 2001 y en Europa en el 2002. Hasta el momento, las autoridades han aprobado el uso de bosentan sólo para pacientes con HAP en clase funcional II/III (Europa) o III/IV (USA y Canadá). Se han realizado varios ensayos clínicos con este fármaco y cada vez es mayor la experiencia clínica en distintos tipos de HP17.
Hipertensión arterial pulmonar idiopática y asociada a esclerodermia
La eficacia de bosentan fue demostrada por primera vez en 2001, en un estudio piloto fase II realizado por Channick y col.18 que incluyó 32 pacientes en clase III con HAP idiopática o asociada a enfermedad de colágeno (AC-351). Fueron randomizados 2:1 para recibir 125 mg/12 hs de bosentan o placebo. A las 12 semanas se objetivó una mejoría significativa en los parámetros hemodinámicos: aumento del índice cardíaco y descenso de la presión auricular derecha y de la resistencia vascular pulmonar (Figura 6). Asimismo mejoró la capacidad funcional, evidenciado por un aumento del test de 6 minutos (+ 60 metros).

Figura 6. Aspectos fisiopatológicos del síndrome de Eisenmenger. IC: índice cardíaco. RVP: resistencia vascular pulmonar.
En un ensayo clínico siguiente fase III (BREATHE-1: Bosentan Randomized Trial of Endothelin Antagonist THErapy-1), bosentan mejoró la distancia caminada en 6 minutos en 44 metros después de 16 semanas de tratamiento, además de mejorar significativamente el índice de disnea de Borg, la clase funcional y el tiempo de empeoramiento clínico19. Se incluyeron 213 pacientes en clase III-IV de la New York Heart Association (NYHA), los que fueron randomizados 1:1 a recibir placebo o bosentan a dosis 62,5 mg/12 hs durante 1 mes seguidos de 125 mg/12 hs o 250 mg/12 hs durante 12 semanas. Ambas dosis de bosentan produjeron un efecto beneficioso, aunque fue más pronunciado en la dosis de 250 mg/12 hs respecto a la de 125 mg/12 hs (+ 54 y + 35 metros, respectivamente). Sin embargo, no se pudo establecer una clara relación dosis/respuesta. Hubo diferencias en la respuesta de ambos grupos de pacientes, mientras que en la HAP idiopática la distancia recorrida se incrementó a +46 metros (vs -5 metros en placebo), en la esclerodermia se evitó el deterioro (+ 3 en bosentan vs -40 metros en el placebo).
En cuanto a la hepatotoxicidad, el aumento de las transaminasas de más de 3 veces el límite superior normal fue más severa y con mayor frecuencia en la dosis de 250 mg/12 hs (14%) con respecto a 125 mg/12 hs (4%). No obstante, se observó una reducción de las transaminasas al disminuir la dosis. Es por ello que la dosis recomendada se estableció en 125 mg/12 hs. A la vez que, las autoridades europeas (European Medicines Agency: EMEA) solicitaron la realización de un sistema de vigilancia postcomercialización para valorar en forma más exacta los efectos hepatotóxicos observados (TRAX PMS), como veremos más adelante.
El estudio BREATHE-1 incluyó un subestudio ecocardiográfico en 85 pacientes con HAP idiopática20. El índice cardíaco estimado por Doppler, el índice de Tei, la relación de área diastólica entre el VD/VI (ventrículo izquierdo) y la escala de graduación del derrame pericárdico evolucionaron favorablemente en los pacientes con bosentan versus placebo.
En los estudios abiertos a continuación con bosentan, 169 pacientes con HAP idiopática enrolados en los 2 estudios previos (AC-351 y Breathe-1) fueron seguidos durante una media de 2,1 ± 0,5 años21. Se observó una tasa de sobrevida de 96 y 89% al año y dos años respectivamente (versus el 69 y 57% de la cohorte de referencia de la NIH, basada en parámetros hemodinámicos). Los predictores de mal pronóstico fueron la clase funcional IV de la NYHA y una distancia recorrida en el test de 6 minutos menor a 358 m en la evaluación inicial. A su vez, el 85 y 70% de los pacientes permanecían estables con bosentan como monoterapia al cabo del primer y segundo año, respectivamente. Se produjeron 20 muertes y 3 trasplantes pulmonares, y el 19% de pacientes recibieron tratamiento combinado con prostanoides. Se produjo una elevación patológica de las transaminasas en un 14,9% de los pacientes, siempre reversible y en el 7,2% la elevación fue superior a 5 veces el límite superior normal. Se realizó un subestudio en la población con HP asociada a la esclerodermia (66 pacientes, 44 con bosentan y 22 placebo), observándose una sobrevida del 85,9% al año y del 73,4% a los 2 años22. Estas tasas de sobrevida son significativamente mayores a las históricas de pacientes con HAP asociada a esclerodermia y no tratados, en donde sobrevivían 45% al año y 35% a los 2 años23. Ocho pacientes (16%) debieron recibir epoprostenol adicional a bosentan y 7 pacientes (14%) recibieron epoprostenol en sustitución a bosentan. Por último, Denton y col. realizaron un análisis a largo plazo de 53 pacientes con HAP asociada a conectivopatías en clase funcional III y que recibieron bosentan 62,5 mg/12 hs en las primeras 4 semanas y luego 125 mg/12 hs en las siguientes 44 semanas, completando 1 año de seguimiento24. Encontraron que un 27% de los pacientes mejoraban la clase funcional y un 16% la empeoraban al año, con un 92% de sobrevida al año y 68% sin empeoramiento clínico. Recientemente, se realizó un ensayo doble ciego, aleatorizado y controlado con placebo, diseñado específicamente para estimar los efectos de bosentan en pacientes con HAP en clase funcional II (EARLY: Endothelin Antagonist tRial in miLdlY symptomatic PAH patients)25. El análisis se realizó con 168 pacientes en clase funcional II (80 recibieron bosentan: 62,5 mg/12 hs en las primeras 4 semanas y 125 mg/12 hs en las siguientes 20 semanas y 88 placebo) que completaron un período de seguimiento de 6 meses. Se incluyeron pacientes del grupo I es decir con HAP idiopática, familiar, asociada a VIH, CIA con defecto menor a 2 cm, CIV con defecto menor a 1 cm, conducto arterioso persistente o asociada a conectivopatías y enfermedades autoinmunes.
Los principales hallazgos fueron una caída significativa de la resistencia vascular pulmonar (22,6%, p<0,0001) con una mejoría no significativa del test de la marcha de 6 min (19,1 m; p=0,0758) y una disminución significativa del tiempo al empeoramiento clínico del 77% comparado con el placebo (log rank p=0,0114). Como objetivo secundario observaron una caída significativa del NTproBNP a los 6 meses (-471 ng/ml; p=0,0003). Los autores concluyen que los pacientes con HAP en clase funcional II también empeoran sin tratamiento, y este empeoramiento ya lo podemos ver a los 6 meses (evolución del grupo placebo) y que el tratamiento con bosentan puede retrasar de forma significativa el deterioro clínico de estos pacientes.
Por último, cabe señalar que en un reciente estudio retrospectivo de 103 pacientes con HAP idiopática en clase funcional III (91) y clase IV (12), Provencher y col. confirman los hallazgos descritos en los estudios aleatorizados descritos previamente26. A los 4 meses de tratamiento con bosentan, la distancia recorrida en 6 minutos aumentó significativamente (322 ± 15 a 364 ± 109 metros) con mejoría de los parámetros hemodinámicas, mejoría que se mantuvo hasta el año de tratamiento. La tasa de sobrevida global estimada fue de 89% a los dos años. De las variables asociadas a la evolución a largo plazo se encontró que la distancia recorrida en 6 minutos y la presión auricular derecha basal y el porcentaje de cambio de la resistencia vascular pulmonar a los 4 meses son las más significativas.
Hipertensión pulmonar asociada a cardiopatías congénitas
Los defectos septales auriculares y ventriculares, así como defectos cardíacos complejos pueden desarrollar HAP. Si estos defectos permanecen sin reparación, se mantiene un cortocircuito izquierda-derecha, con un aumento del flujo sanguíneo hacia la circulación pulmonar. Con el paso del tiempo la luz de las arteriolas disminuyen, debido en parte a la proliferación de los múltiples componentes de la pared vascular inducida en parte por el aumento de la tensión de cizallamiento17. Estos cambios se asocian con disfunción endotelial y muscular determinando un aumento persistente de la resistencia vascular pulmonar. En cuanto la PMAP aumenta, el cortocircuito se invierte (de derecha a izquierda), produciéndose desoxigenación de la sangre arterial lo cual se manifiesta clínicamente con la aparición de cianosis labial y subungueal (Figura 7). Este fenómeno de cortocircuito derecha-izquierda con resistencias vasculares pulmonares elevadas define el síndrome de Eisenmenger (SE) y representa una HAP particularmente severa, con pobre tolerancia al ejercicio y mala calidad de vida, aunque con una mayor sobrevida comparada con las otras formas de HAP. Ello es debido al fenotipo fetal que persiste en el ventrículo derecho, pudiendo soportar resistencias pulmonares de 2800 a 3400 dinas.s.cm-5. Las características histopatológicas del SE son groseramente idénticas a las observadas en otras formas de HAP. Asimismo se han encontrado niveles de ET-1 en estos pacientes, aunque sin una correlación tan clara con la severidad hemodinámica. Ello puede deberse a múltiples factores como el tamaño y localización del defecto y la 'susceptibilidad individual'27. Estos hallazgos fueron la base del ensayo BREATHE-5, doble ciego, aleatorizado y controlado con placebo en pacientes con SE, seguidos 16 semanas28. El primer objetivo fue demostrar que bosentan no agravaba el shunt y por ende no deterioraba la saturación arterial de oxígeno. Concomitantemente se produjo una reducción significativa de la resistencia vascular pulmonar (-472 dinas.s.cm-5; p=0,04), con un aumento de la distancia recorrida en 6 min (+53,1 m, p=0,008) y mejora de la clase funcional NYHA.

Figura 7. Evolución de shunt izquierda-derecha en la hipertensión arterial pulmonar.
En un estudio abierto continuado (BREATHE-5 OLE), Gatzoulis y col. comunican un mantenimiento de la mejora en la capacidad funcional al cabo de 40 semanas en los pacientes que seguían recibiendo bosentan29. A su vez, los pacientes del grupo placebo que en la fase abierta del estudio recibieron bosentan mejoraron la distancia caminada a 32 m.
Hipertensión pulmonar asociada a virus de la inmunodeficiencia humana
El ensayo clínico BREATHE-4 fue diseñado como un estudio prospectivo y no comparativo de 16 semanas de duración. Se incluyeron 16 pacientes con HP asociada a infección VIH, 15 en clase III de la NYHA y uno en clase IV; todos excepto 1 recibían simultáneamente tratamiento con una combinación de al menos 3 fármacos antirretrovirales30. En la semana 16, todos los parámetros de eficacia del estudio mejoraron: el perfil hemodinámico (índice cardíaco +0,9±0,7 l/min/m2 y la resistencia vascular pulmonar -339 ± 209 dinas.s.cm2), el test de 6 min (91 ± 60 m) y las variables ecocardiográficas (reducción del área de la aurícula y del ventrículo derechos, del derrame pericárdico y del índice de excentricidad diastólico). Durante el estudio no hubo ningún muerto y ningún paciente requirió tratamiento de rescate. Se produjo un 12% de elevación de transaminasas que no requirieron la retirada de bosentan. El tratamiento con bosentan no tuvo impacto negativo en el control de la infección VIH.
Hipertensión pulmonar idiopática y asociada a cardiopatía congénita en niños
El ensayo clínico BREATHE-3 fue diseñado como un estudio abierto, prospectivo, múltiple-dosis y no controlado con placebo de 12 semanas de duración, con el objetivo de estudiar la farmacocinética, eficacia y seguridad de bosentan en niños con HP. Se incluyeron 19 niños entre 4 y 17 años, 10 con HAP idiopática y 9 asociada a cardiopatía congénita, en clase funcional III de la NYHA31. Se dividieron en 3 grupos según peso y el uso simultáneo de epoprostenol (52% de los pacientes). La dosis administrada en las primeras 4 semanas fue de 62,5 mg/12 hs (> 40 kg), 31,5 mg/12 hs (20-40 kg) y 31,5 mg/día (10-20 kg). La dosis de mantenimiento fue de 125 mg/12 hs, 62,5 mg/12 hs y 31,5 mg/12 hs, respectivamente. En la semana 12 se produjo una mejora significativa del perfil hemodinámica con reducción de las resistencias vasculares pulmonares. En 3 pacientes, se elevaron las transaminasas y en 2 se suspendió el tratamiento con bosentan. No hubo exitus durante el estudio.
Hipertensión pulmonar tromboembólica crónica (HPTEC)
El primer estudio doble-ciego, aleatorizado y controlado por placebo que analizó los efectos de bosentan en la hemodinamia y la capacidad funcional en pacientes con HPTEC es el estudio BENEFiT (Bosentan Effects in iNopErable Forms of chronic Thromboembolic pulmonary hypertension)32. Se incluyeron 157 pacientes, randomizados: 80 placebo y 77 recibieron bosentan ya sea con HPTEC inoperable o con HP persistente luego de 6 meses de la cirugía. Los objetivos co-primarios fueron el cambio en la resistencia vascular y la distancia recorrida en 6 min. Se observó una reducción significativa en la resistencia vascular (-24,1%, -193 dinas.s.cm-5; p=0,0001) y en el índice cardíaco (+0,3 l/min/m2; p=0,0007), con una reducción del NT-ProBNP (-622 ng/l; p=0,0034). Sin embargo, no hubo cambios significativos en el test de la marcha. Una de las hipótesis que podría explicar estas discrepancias en los resultados es que los pacientes con HPTEC son más añosos y que tomaría más de 16 semanas para que la mejoría hemodinámica se expresara en una mejoría clínica17. Estos resultados sugieren que bosentan mejora la hemodinámica en los pacientes con HPTEC inoperable o con HP persistente después de la endarterectomía pulmonar.
Hipertensión porto-pulmonar
En la hipertensión porto-pulmonar (HPP) no existen ensayos clínicos que evalúen la eficacia y seguridad de bosentan, aunque existen experiencias preliminares que muestran que la medicación fue bien tolerada y no hubo evidencia de daño hepático relacionado con el uso de bosentan33. Hoeper y col. valoraron el tratamiento con bosentan en 11 pacientes con clase funcional III-IV, con cirrosis hepática estadio A de Child y con HPP severa. Todos los pacientes mejoraron su capacidad funcional tras 1 año de tratamiento (test de 6' de 310 ± 102 a 388 ± 81 metros y consumo pico de oxígeno de 12,6 ± 3,5 a 16,6 ± 2,8 ml/kg/min), conjuntamente con el perfil hemodinámico dado por la reducción de las resistencias vasculares pulmonares. La HPP moderada severa (PAPM > 35 mm Hg; resistencia vascular pulmonar > 3,1 UW) supone un elevado riesgo perioperatorio para el paciente candidato a trasplante hepático, pudiendo alcanzar una mortalidad perioperatoria > 50%. Clift y col. comunican la evolución favorable de un paciente con HPP moderada que se incrementó tras el trasplante, determinando una insuficiencia cardíaca derecha que mejoró con el agregado de bosentan a la terapia de base34.
Tratamientos combinados
Un esquema terapéutico basado en la combinación de fármacos con diferentes mecanismos de acción podría obtener mejores resultados que la monoterapia. Existen 2 posibles tácticas de tratamiento combinado: a) la combinación secuencial, en donde se agrega un segundo fármaco al que venía recibiendo el paciente, y b) inicio concomitante de 2 fármacos. Bosentan ha sido estudiado en combinación con otras drogas específicas para el tratamiento de la HP.
1. Bosentan combinado con prostanoides
Se ha realizado con 2 pautas diferentes. En pacientes tratados crónicamente con prostanoides que comienzan a deteriorar su situación clínica, en los que el tratamiento combinado es una estrategia de rescate. Se analizó una cohorte de 20 pacientes tratados crónicamente con iloprost o beraprost (16 ± 13 meses) con mala evolución, a los que se añadió bosentan a dosis habituales35. Se observó una mejoría de la capacidad funcional a los 3 meses de tratamiento combinado dado por el test de 6 minutos y el consumo pico de oxígeno en la ergoespirometría. Recientemente, se han comunicado experiencias inversas, en donde el paciente que estaba recibiendo bosentan, se le agregaba iloprost inhalado. Dos estudios aleatorizados y controlados, el STEP-1 (Safety and pilot efficacy Trial in combination with bosentan for evaluation in Pulmonary arterial hypertension) y el COMBI (Combination Therapy of Bosentan and Aerolised Iloprost in idiopathic Pulmonary Arterial Hypertension), han demostrado resultados mixtos. Ambos confirman que la combinación es segura, pero sólo el STEP-1 demostró una mejoría clínica36,37.
Recientemente, se comunicó un estudio retrospectivo de 38 pacientes con HAP de un único centro tratados con treprostinil s/c en forma crónica, 2 en clase II, 29 en clase III y 7 en clase IV de la NYHA38. Bosentan se agregó en 19 pacientes, de los cuales 7 tenían efectos colaterales intolerables al treprostinil (duración media de 770 ± 307 días y 38 ± 18 ng/kg/min al inicio de bosentan). El seguimiento con terapia combinada fue de 485 días. La combinación treprostinil + bosentan aumentó la distancia recorrida en 6' (333 ± 79 vs 374 ± 110 metros), y redujo la PMAP y la presión de la aurícula derecha. Doce pacientes estaban en clase II, 5 en clase III y uno en clase I. No existieron efectos colaterales y la dosis de treprostinil se mantuvo después de agregar bosentan.
La segunda estrategia plantea el tratamiento combinado como estrategia terapéutica inicial intentando lograr su máxima eficacia en el período más breve de tiempo. Así se llevó a cabo el estudio BREATHE-2, ensayo clínico randomizado y controlado con placebo de 16 semanas de duración que comparó la eficacia y seguridad de epoprostenol + placebo frente a epoprostenol + bosentan como tratamiento inicial en 33 pacientes con HAP idiopática (80%) y colagenosis (20%), en clase funcional III (76%) y IV (24%) de la NYHA39.
Desafortunadamente este estudio no demostró beneficios significativos de la rama epoprostenol + bosentan, entre las causas figuran el bajo número de pacientes y el corto seguimiento.
2. Bosentan combinado con sildenafilo
Estudios preliminares sugieren que bosentan puede ser combinado en forma segura con el sildenafilo, un inhibidor de la fosfodiesterasa-5 en pacientes con HAP. Dos ensayos no controlados describen una mejoría en la capacidad de ejercicio (distancia recorrida y consumo pico de oxígeno) cuando el sildenafilo se agregaba a bosentan en pacientes con HAP idiopática40. Dicho efecto beneficioso se mantuvo durante un tiempo medio de seguimiento de 9 meses. Sin embargo, no hubo un claro beneficio en pacientes con HAP asociada con esclerodermia, sugiriendo que no todas las formas de HAP responden de la misma forma a la terapia médica41.
Un elemento a tener en cuenta cuando se combina bosentan + sildenafilo es la interacción farmacocinética entre ambos fármacos, resultando en un aumento de la concentración de bosentan y un descenso de la de sildenafilo, lo que podría aumentar la hepatotoxicidad, a la vez de reducir la eficacia del tratamiento combinado. Si bien hasta ahora no hay evidencia de un aumento de hepatotoxicidad por el tratamiento combinado, sí se ha demostrado una disminución en la concentración plasmática del sildenafilo42, aunque si ello se asocia con una menor eficiencia clínica del tratamiento combinado, es una cuestión debatida.
El estudio de la combinación de bosentan y sildenafilo versus sildenafilo en monoterapia (Combination Of Bosentan and Sildenafil versus Sildenafil Monotherapy on morbidity and mortality in symptomatic PAtientS with PAH: COMPASS-1) demostró que la respuesta hemodinámica aguda a sildenafilo fue similar en pacientes pretratados con bosentan y los no tratados, pero ello no es suficiente para excluir la hipótesis de que bosentan disminuye la eficacia terapéutica de sildenafilo (datos no publicados).
Actualmente, está en curso el estudio COMPASS-2 que examinará los efectos a largo plazo del agregado de bosentan a sildenafilo9. Incluirá unos 600 pacientes con HAP que reciben por lo menos durante 12 semanas o más sildenafilo a dosis ≥ 20 mg, a los que se agregará durante las primeras 4 semanas bosentan a 62,5 mg/12 hs y luego 125 mg/12 hs como dosis de mantenimiento. Como objetivos co-primarios se valorará el test de los 6' a las 16 semanas y la morbimortalidad con un análisis farmacoeconómico concomitante.
Tolerancia y perfil de seguridad
En los distintos ensayos clínicos se ha constatado que bosentan puede producir hepatotoxicidad, especialmente a dosis altas. Este efecto puede observarse en las fases precoces o tardías del tratamiento, aunque generalmente ocurren en los primeros 6 meses de iniciado el tratamiento. Estas anomalías son generalmente asintomáticas y se resuelven al disminuir la dosis o suspender el tratamiento. El mecanismo más probable de toxicidad hepática es la competición dosis dependiente de bosentan y sus metabolitos con la excreción biliar de las sales biliares, produciendo una retención de las sales biliares que pueden ser tóxicas para los hepatocitos43. Inicialmente los datos de tolerabilidad de bosentan fueron aportados por el fabricante a partir del análisis por un lado de los 245 pacientes adultos provenientes de los 2 primeros ensayos (AC-351 y BREATHE-1) y por otro del estudio BREATHE-3 para la población pediátrica. No obstante y en forma reciente, se aportaron datos adicionales a partir de un programa de vigilancia postcomercialización, el TAP (Tracleer Access Program) en EUA y el TRAX PMS (Tracleer Excellence PostMarketing Surveillance) en 18 países de Europa44. Este último registró la incidencia de efectos adversos en casi 4848 pacientes adultos (> 12 años) con HAP (80% de los pacientes tratados en Europa, de mayo 2002 a noviembre 2004) que recibieron bosentan durante una media de 31 semanas45, y 146 niños entre 2 y 12 años que recibieron bosentan durante una media de 29 semanas46. En los adultos se observó un aumento mayor a 3 veces el límite superior normal de las transaminasas en un 7,6% de tasa cruda, correspondiente a una tasa anualizada de 10,1%, muy similar a las tasas reportadas en los ensayos (12,8% y 11,2%). De los 380 pacientes con hepatotoxicidad, 38% continuaron con bosentan, 13% reintrodujeron bosentan (de los cuales 14 tuvieron que discontinuar) y 49% discontinuaron. Ninguno presentó insuficiencia hepática y todos revirtieron sin secuelas entre algunos días y 9 semanas ya sea espontáneamente o luego de descender la dosis o de discontinuarlo44. No hubo diferencias significativas en función de la etiología de la HAP. A nivel pediátrico, predominó la etiología idiopática (40,4%) y la HAP asociada a cardiopatías congénitas (45,2%), con una tasa cruda de hepatotoxicidad de 2,7% y una tasa de discontinuación de 14,4%, lo que sugiere una mayor tolerancia de los niños a bosentan46. En cuanto al control periódico de las enzimas, se recomienda al inicio del tratamiento, a los 15 días y 30 días de iniciado el tratamiento, y luego en forma mensual. En la Tabla 3, se resume el manejo clínico frente al aumento de las transaminasas.

Bosentan está contraindicado en el embarazo por sus efectos teratogénicos. Interacciona con los anticonceptivos hormonales orales, disminuyendo su eficacia, y con la glibenclamida, incrementando el riesgo de toxicidad hepática.
En los ensayos clínicos, se ha observado anemia en un reducido número de pacientes, en general ligera, reversible y de naturaleza desconocida (no hay toxicidad medular ni hemólisis), siendo recomendable verificar periódicamente la hemoglobina y el hematocrito. Además, se ha observado retención de líquidos y edemas en las extremidades inferiores, cefaleas y sofocos44. Existe inquietud de que los antagonistas de la endotelina puedan causar atrofia testicular e infertilidad en el varón como efecto clase, por lo que los varones en edad fértil deben conocer esta posibilidad antes de iniciar la medicación.
Conclusiones
El antagonista dual no selectivo de los receptores de ET-1, bosentan, es un eficaz tratamiento en la HP:
- Mejora en la capacidad de ejercicio.
- Mejora en la clase funcional y calidad de vida.
- Mejora de la hemodinamia cardiopulmonar.
- Retrasa el deterioro clínico y aumenta la supervivencia.
- Muestra aumento de la supervivencia en el análisis restrospectivo de los estudios de extensión.
Teniendo en cuenta las recomendaciones de uso, bosentan ha sido aprobado para el tratamiento de los pacientes con HP en clase funcional III y IV de la NYHA en EUA y Canadá. En Europa, la Agencia Europea del Medicamento (EMEA), ha aprobado bosentan para el tratamiento de los pacientes en clase II a III de la NYHA, especificando que la eficacia del tratamiento ha sido demostrada en pacientes con HP idiopática y asociada a colagenosis sin fibrosis pulmonar significativa.
Con el nivel de evidencia actual, las recomendaciones en Europa son:
- Clase I; nivel de evidencia A para la HAP idiopática y la HAP asociada a colagenosis sin fibrosis pulmonar significativa en clase II y III de la NYHA.
- Clase IIa; nivel de evidencia C para la HAP idiopática y la HAP asociada a colagenosis sin fibrosis pulmonar significativa en clase IV de la NYHA.
1. Gómez-Sánchez MA. Hipertensión Pulmonar. Editorial Ergon. Madrid, España; 2008. [ Links ]
2. Hatano S, Strasser T. Primary pulmonary hypertension. Report of the WHO Meeting. Geneve: World Health Organization; 1975. [ Links ]
3. Rich S, Dantzker DR, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Primary pulmonary hypertension. A National Prospective Study. Ann Intern Med 1987;107:216-23. [ Links ]
4. Rich S, editor. Executive Summary from the World Symposium on Primary Pulmonary Hypertension 1998 co-sponsored by the World Health Organization. (http:/www.who.int/ncd/cvd/pph.html). [ Links ]
5. Galié N, Torbicki A, Barst R, Dartevelle P, Haworth S, Higenbottam T, et al. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 2004;25:2243-78. [ Links ]
6. Barst R, Rubin LJ. 4th World Symposium on Pulmonary Hypertension. February 11-14, 2008. Dana Point, California. [ Links ]
7. Dupuis J. Endothelin: setting the scene in pulmonary arterial hypertension. Eur Respir Rev 2007;16:3-7. [ Links ]
8. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Eng J Med 1993;328:1732-9. [ Links ]
9. Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur Respir J 2008;31:407-15. [ Links ]
10. Rubens C, Ewert R, Halank M, Wensel R, Orzechowski HD, Schultheiss H-P, et al. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest 2001;120:1562-69. [ Links ]
11. Galié N. Eur J Clin Invest 1996; 26(Suppl 1): 273. [ Links ]
12. Migneault A, Sauvageau S, Villeneuve L, Thorin E, Fournier A, Leblanc N, et al. Chronically elevated endothelin levels reduce pulmonary vascular reactivity to nitric oxide. Am J Respir Crit Care Med 2005;171:506-13. [ Links ]
13. Hocher B, Schwartz A, Fagan KA, Thone-Reineke C, El- Hag K, Kusserow H. et al. Pulmonary fibrosis and chronic lung inflammation in ET-1 transgenic mice. Am J Respir Cell Mol Biol 2000;23:19-26. [ Links ]
14. Bauer M, Wilkens H, Langer F, Schneider SO, Lausberg H, Schafers HJ. Selective up regulation of endothelin B receptor gene expression in severe pulmonary hypertension. Circulation 2002;105:1034-34. [ Links ]
15. Langleben D, Dupuis J, Langleben I, Hirsch AM, Baron M, Senécal J-L, et al. Etiology-specific endothelin-1 clearance in human precapillary pulmonary hypertension. Chest 2006;129:689-95. [ Links ]
16. Oldfield V, Lyseng-Williamson KA. Bosentan: a review of its use in pulmonary arterial hypertension and systemic sclerosis. Am J Cardiovasc Drugs 2006;6:189-208. [ Links ]
17. Humbert M. Dual endothelin receptor antagonism: setting Standard in PAH. Eur Respir Rev 2007;16:13-18. [ Links ]
18. Channick RN, Simonneau G, Sitbon O, Robins IM, Frost A, Tapson VF, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomized placebo-controlled study. Lancet 2001;358:119-23. [ Links ]
19. Rubin LJ, Badesch D, Barst R, Galie N, Black C, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension (for the 'Bosentan Randomized Trials of Endothelin Antagonist Therapy': BREATHE Study Group). N Eng J Med 2002;346:896-903. [ Links ]
20. Galié N, Hinderliter A, Torbicki A, Fourme T, Simonneau G, Pulido T, et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic and Doppler measures in patients with primary pulmonary hypertension. J Am Coll Cardiol 2003;41:1380-6. [ Links ]
21. McLaughlin VV, Sitbon O, Badesch DB, Barst RJ, Black C, Galié N, et al. Survival with first-line bosentan in patients with primary pulmonary hypertension. Eur Respir J 2005;25:244-9. [ Links ]
22. Denton CP, Humbert M, Rubin L, Black CM. Bosentan treatment for pulmonary arterial hypertension related to connective tissue disease: a subgroup analysis of the pivotal clinical trials and their open-label extensions. Ann Rheum Dis 2006:65:1336-40. [ Links ]
23. Koh E, Lee P, Gladman D, Abu-Shakra M. Pulmonary hypertension in systemic sclerosis: an analysis of 17 patients. Rheumatology 1996;35:989-93. [ Links ]
24. Denton CP, Pope JE, Meter HH, Gabrielli A, Boonstra A, van den Hoogen FHJ, et al. Long-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseases. Ann Rheum Dis 2008;67:1222-8. [ Links ]
25. Galié N, Rubin LJ, Hoeper MM, Jansa P, Al-Hiti H, Meyer GMB, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind randomised controlled trial. Lancet 2008;371:2093-100. [ Links ]
26. Provencher S, Sitbon O, Humbert M, Cabrol S, Jais X, Simonneau G. Long-term outcome with the first-line bosentan therapy in idiopathic pulmonary arterial hypertension. Eur Heart J 2006;26:589-95. [ Links ]
27. Cacoub P, Dorent R, Maistre G, Nataf P, Carayon A, Piette C, et al. Endothelin-1 in primary pulmonary hypertension and the Eisenmenger syndrome. Am J Cardiol 1993;71:448-50. [ Links ]
28. Galié N, Beghetti M , Gatzoulis MA, Granton J, Berger RM, Lauer A, et al. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double bind, randomized, placebo-controlled study. Circulation 2006;114:48-54. [ Links ]
29. Gatzoulis MA, Beghetti M, Galié N, Granton J, Berger RM, Lauer A, et al. Long-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension study. Int J Cardiol 2008;127:27-32. [ Links ]
30. Sitbon O, Gressin V, Speich R, MacDonald PS, Opravil M, Cooper DA, et al. Bosentan for the treatment of human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med 2004;140:1212-7. [ Links ]
31. Barst RJ, Ivy D, Dingemanse J, Wildlitz A, Schmitt K, Doran A, et al. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin Pharmacol Ther 2003;73:372-82. [ Links ]
32. Hughes RJ, Jais X, Bonderman D. The efficacy of bosentan in inoperable chronic thromboembolic pulmonary hypertension: a 1-year follow-up study. Eur Respir J 2006;28:138-43. [ Links ]
33. Hoeper MM, Halank M, Marx C, Hoefken G, Seyfarth HJ, Schauer J, et al. Bosentan therapy for portopulmonary hypertension. Eur Respir J 2005;25:502-8. [ Links ]
34. Clift PF, Towend JN, Bramhall S, Isaac JL. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004;77:1774-5. [ Links ]
35. Hoeper MM, Taha N, Bekjarova A, Gatzke R, Spiekerkoetter E. Bosentan treatment n patients with primary arterial hypertension receiving non-parenteral prostanoids. Eur Respir J 2003;22:330-4. [ Links ]
36. Hoeper MM, Leuchte H, Halank M, Wilkens H, Meyer FJ, Seyfarth HJ, et al. Combining inhaled iloprost with bosentan in patients with idiopathic pulmonary arterial hypertension. Eur Respir J 2006;28:691-94. [ Links ]
37. McLaughlin VV, Oudiz RJ, Frost A, Tapson VF, Murali S, Channick RN, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 2006:174:1257-63. [ Links ]
38. Benza RL, Rayburn BK, Tallaj JA, Pamboukian SV, Bourge RC. Treprostinil-based therapy in the treatment of moderate-to-severe pulmonary arterial hypertension. Chest 2008;134:139-45. [ Links ]
39. Hoeper MM, Barst RJ, Robbins I, Cannick R, Galié N, Boonstra A, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J 2004;24:353-9. [ Links ]
40. Hoeper MM, Faulenbach C, Golpon H, Winkler J, Welte T, Niedermeyer J. Combination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertension. Eur Respir J 2004;24:1007-10. [ Links ]
41. Mathai SC, Girgis RE, Fischer MR, Champion HC, Houston-Harris T, Zaiman A, Hassoun PM. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J 2007;29:469-75. [ Links ]
42. Paul GA, Gibbs JS, Boobis AR, Abbas A, Wilkins MR. Bosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertension. Br J Clin Pharmacol 2005;60:107-112. [ Links ]
43. Fattinger K, Funk C, Pantze M, Weber C, Reichen J, Steiger B, Meier PJ. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001;69:223-31. [ Links ]
44. Segal ES, Valette C, Oster L, Bouley L, Edfjall C, Herrmann P, et al. Risk management strategies in the postmarketing period. Safety experience with US and European Bosentan surveillance programmes. Drug Safety 2006;28:971-80. [ Links ]
45. Humbert M, Segal ES, Kiely DG, Carlsen J, Schwierin B, Hoeper MM. Results of European post-marketing surveillance of bosentan in pulmonary hypertension. Eur Respir J 2007;30:338-44. [ Links ]
46. Beghetti M, Hoeper MM, Kiely DG, Carlsen J, Schwierin B, Segal ES, et al. Safety experience with bosentan in 146 children 2-11 years old with pulmonary arterial hypertension: results from the European Postmarketing Surveillance Program. Pediatric Res 2008;64:200-4. [ Links ]

