










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. v.4 n.1 Ciudad Autónoma de Buenos Aires ene./mar. 2009
SIMPOSIO INTERNACIONAL DE HIPERTENSIÓN PULMONAR
Presentación clínica y clasificación actual de la hipertensión arterial pulmonar
Guillermo Bortman*
* Médico cardiólogo. Jefe de Trasplantes. Servicio de Cardiología. Sanatorio de la Trinidad-Mitre. Ciudad de Buenos Aires. República Argentina.
Coordinador de Trasplantes del Sanatorio “Denton Cooley”. Ciudad de Buenos Aires. República Argentina.
Coordinador de Trasplantes del Hospital Italiano. Ciudad de Mendoza. Mendoza. República Argentina.
Director de Cardiología del Htal. de Alta Complejidad Médica. Ciudad de Formosa. Formosa. República Argentina.
Director de Emprendimientos de Salud y Subdirector Médico del Club Boca Junior. Ciudad de Buenos Aires. República Argentina.
Correspondencia: Dr. Guillermo Bortman
Departamento Cardiovascular Sanatorio de la Trinidad-Mitre.
Bartolomé Mitre 2553 Piso 1º - CP: 1039 - Ciudad de Buenos Aires. República Argentina.
E-mail: gbortman@fibertel.com.ar
Recibido: 26/08/2008
Aceptado: 12/01/2009
Palabras clave: Hipertensión arterial pulmonar; Signos y síntomas; Clasificación; Diagnóstico
En esta comunicación analizaremos, básicamente, la presentación clínica de los pacientes portadores de hipertensión arterial pulmonar (HAP), y cómo se puede evitar llegar al diagnóstico en fases muy avanzadas. Esto es importante, ya que si bien sigue siendo una enfermedad rara, frecuentemente, se encuentra subdiagnosticada. Aún cuando, en este momento, tenemos un mayor conocimiento de su fisiopatología.
Por supuesto que todavía existe el diagnóstico por exclusión. Habitualmente cuando el paciente llega a la consulta, en general, se encuentra en la etapa final de su enfermedad; lo cual no nos posibilita detener la enfermedad en una fase temprana y así tomar una actitud terapéutica precoz que mejore la sobrevida de estos pacientes.
Pero también, contamos en la actualidad con una terapéutica mucho más específica respecto a los mecanismos fisiopatológicos de la HAP. Es por ello que tenemos muy buenas expectativas y avanzadas investigaciones que dan evidencia de que nos encontramos en las etapas iniciales de la terapéutica de esta enfermedad tan lamentable.
En un registro francés (Figura 1), el 75% de los pacientes llegan a la consulta en clase funcional (CF) III-IV1. Y utilizando, un dicho que decimos habitualmente “hasta mi tía diagnostica una hipertensión pulmonar en esta situación”.

Figura 1. Datos clínicos y hemodinámicos para el diagnóstico de la hipertensión arterial pulmonar. Resultados de un registro nacional francés (2006)1.
También es importante mencionar este estudio para desmitificar lo relativo a la edad. Ya no es una patología que afecta a mujeres entre los 20 y los 30 años, sino que entre 674 pacientes, la edad media fue de 50 años.
Con respecto a la sintomatología de la HAP, tenemos que decir que los síntomas son absolutamente ambiguos (Tabla 1). Si mencionamos algunos de ellos como: disnea, fatiga, dolor de pecho, síncope o pre-síncope y edema, nos encontramos con que hay infinidad de patologías capaces de producirlos en forma similar, por lo tanto, no todo paciente que padezca estos síntomas va a padecer una HAP. Pero es evidente que no estamos acostumbrados a diagnosticarla, y entonces la subdiagnosticamos.

Recién en las etapas más avanzadas es el momento en que se empieza a sospechar de ella, pues la presentación es más florida e importante, y es cuando el paciente se hace refractario y no responde a las terapéuticas básicas de la insuficiencia cardíaca (Tabla 2).

En un conocido club de fútbol de la Argentina, hemos hecho un examen en el cual se realiza un screening a toda la población deportiva (1700 atletas, aproximadamente). El examen consiste, básicamente, en la realización de un electrocardiograma (ECG), una telerradiografía (Rx) de tórax y la rutina de laboratorio, y de existir algún tipo de dudas, se completa el examen con un ecocardiograma.
Esto se realiza, no sólo para diagnosticar HAP; sino también, para poder predeterminar otros tipos de patologías como las cardiopatías congénitas, los síndromes de preexcitación (relativamente frecuente en atletas jóvenes de todas las edades), etc. El objetivo es detectar mediante un examen muy sencillo el diagnóstico precoz de estas patología y desde ya permitir un tratamiento más eficiente, y así poder mejorar la evolución de un paciente en CF I o II, cuando todavía tiene presiones pulmonares no tan altas, y sin tanta insuficiencia cardíaca (IC) derecha.
Así y todo, vemos que la sintomatología, si bien es semejante para todas las entidades que causan HAP, el pronóstico (Figura 2) es distinto si la cardiopatía es congénita, que si la HAP es idiopática o secundaria a una colagenopatía, o asociada al virus de inmunodeficiencia humana (VIH)3, de acuerdo a los registros que estamos obteniendo en la actualidad. Es realmente insólito que a los pacientes que están siendo seguidos por los infectólogos con HIV no se les haga un ecocardiograma de control para verificar si no están desarrollando HAP.

Figura 2. Sobrevida de pacientes con hipertensión arterial pulmonar (HAP)3.
CC: Cardiopatía congénita. CV: Colagenopatía vascular. VIH: Virus de la inmunodeficiencia humana. HAPI: Hipertensión arterial pulmonar idiopática. HPP: Hipertensión porto-pulmonar
Al referirnos al hallazgo de los síntomas mencionados, al examen físico y al screening, es importante para poder determinar aquellos pacientes que tienen antecedentes familiares, aquellos pacientes que han utilizado drogas para no comer, o elementos anorexígenos, o toda persona que esté en situación de riesgo (Tabla 3). Obviamente, se debería hacer un screening para la búsqueda por lo menos de algunos signos4.

No nos referimos a signos tan evidentes como una sobrecarga ventricular derecha severa con clínica de hipertensión pulmonar suprasistémica. Pero es evidente que no se pasa de un ECG normal a uno patológico en dos días. Para poder llegar al ECG de la Figura 3, o a la Rx de tórax de la Figura 4, evidentemente tiene que haber pasado un tiempo. Y cuando llegamos a este diagnóstico, los 2,8 años de sobrevida original promedio se han acortado, pues es evidente que por lo menos varios meses de esta patología no fueron detectados (Tabla 4).
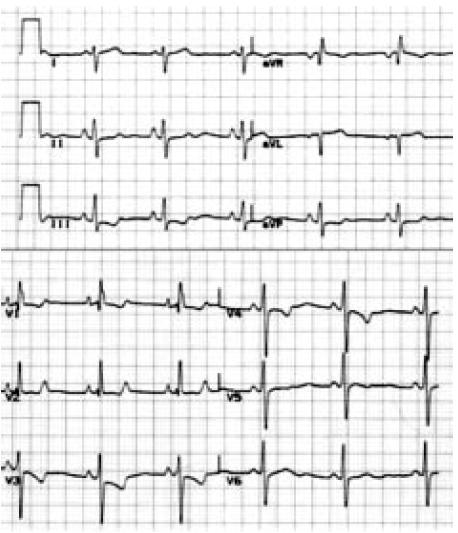
Figura 3. Electrocardiograma de una hipertensión pulmonar.

Figura 4. Telerradiografía de tórax de una hipertensión arterial pulmonar. A) Esquema: P= arterial pulmonar prominente. Q= amputación de flujo periférico. B) Rx de tórax frente.

La resistencia vascular periférica pulmonar aumenta progresivamente, tanto en la etapa asintomática como en la sintomática. Pero sucede en forma diferente con la presión arterial pulmonar. La presión arterial pulmonar también sube, y en un momento determinado empieza a caer. Es muy importante detectarlo, porque cuando ello sucede, probablemente, estemos en presencia de una incipiente fase de IC derecha y claudicación del ventrículo derecho, o sea que los pacientes presentan una evolución desfavorable con signos de IC derecha: hepatomegalia, edemas de miembros inferiores, trastornos de coagulación, agotamiento, aspecto azulado (cianosis), y presencia de cuadros sincopales (Figura 5).

Figura 5. Evolución de la hipertensión arterial pulmonar (deterioro progresivo).
Lo mismo ocurre con el volumen minuto (VM) cardíaco y la presión de la aurícula derecha (AD). El VM en la primera etapa se mantiene para después ir cayendo, y la presión de la AD empieza a subir con agravamiento de los signos de IC derecha.
En conclusión, los síntomas son relativamente ambiguos y solamente en las fases más avanzadas se hacen más evidentes y los elementos clínicos indican que el paciente está teniendo IC derecha. Pero probablemente este momento ya sea tarde para conseguir una terapia eficaz y revertir el cuadro clínico. El mensaje es tratar de buscar los síntomas, o buscar elementos que puedan hacer sospechar lo más precozmente posible, en la forma más anticipada, para tratar que los mecanismos fisiopatológicos que agravan la HAP puedan ser detenidos, al menos parcialmente, para lograr una mayor sobrevida y mejorar la calidad de vida del paciente.
La clasificación que todo el mundo conoce de HAP fue modificada en el 2008, en California, en Dana Point (Tabla 5). La clasificación era relativamente sencilla de entender. Los pacientes con HAP arterial idiopática estaban en el grupo 1. Los pacientes asociados a disfunción ventricular izquierda con IC izquierda, disfunción diastólica, valvulopatías, se encontraban en el grupo 2. Los pacientes con patología pulmonar estaban en el grupo 3: enfermedad pulmonar obstructiva crónica (EPOC), patología intersticial pulmonar, desórdenes del sueño, exposiciones a la altitud. Los pacientes asociados a tromboembolismo pulmonar pertenecen al grupo 4. Y obviamente, las causas desconocidas y mecanismos multifactoriales, o trastornos hematológicos, mieloproliferativos, esplenectomía, vasculitis, sarcoidosis, enfermedades del glucógeno, enfermedad de Gaucher, enfermedades congénitas, eran del grupo 5.

Las modificaciones básicas se refieren al grupo 1, y van a estar relacionadas a las verificaciones de las mutaciones genéticas, específicamente el BMPR2 y el Alk-1, y por supuesto también a causas que no son conocidas aún, y que van a estar incorporadas en los próximos años en estos trastornos y mutaciones genéticas. Se incorporan las drogas y las inducidas por toxinas, y en este caso las asociadas a colagenopatías, infección por HIV, hipertensión portal, los shunts pulmonares, la esquistosomiasis, y las anemias crónicas hemolíticas. En la Tabla 6, se muestran los porcentajes epidemiológicos de esta clasificación.

Conclusión
En resumen, tendríamos que insistir en la búsqueda de esta enfermedad o por lo menos, tratar de descartarla. Un famoso profesor de dermatología, Lois Bachman, decía que la lepra no existe porque nadie la piensa. Y relativamente tenía razón. La lepra existe y cuando uno la busca, la encuentra. Entonces, todos deberíamos pensar que existe esta entidad, y probablemente la encontraremos más frecuentemente. Si a ello le sumamos detectarla con un diagnóstico precoz, considerando los avances que tenemos actualmente y los que vendrán, seguramente exista la posibilidad de disminuir el avance de esta enfermedad que causa estragos en la población, comprometiendo la calidad de vida y la sobrevida.
1. Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, et al. Pulmonary Arterial Hypertension in France: Results from a National Registry. Am J Respir Crit Care Med 2006;173:1023-1030. [ Links ]
2. Rich SA, Dantzker DR, Ayres SM, et al. Primary Pulmonary hypertension: a national prospective study. Ann Intern Med 1987;107:216-223. [ Links ]
3. McLaughlin VV, Presberg KW, Doyle RL, Abman SH, Mc-Crory DC, Fortin T, Ahearn G. Prognosis of pulmonary arterial hypertension. ACCP evidence-based clinical practice guidelines. Chest 2004;126:78S-92S. [ Links ]
4. Galiè N, Torbicki A, Barst R, Dartevelle P, Haworth S, Higenbottam T, Olschewski H, Peacock A, Pietra G, Rubin LJ, Simonneau G. Guidelines on diagnosis and treatment of pulmonary arterial hypertension: The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of Cardiology. Eur Heart J 2004;25:2243-2278. [ Links ]

