










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. v.4 n.1 Ciudad Autónoma de Buenos Aires ene./mar. 2009
SIMPOSIO INTERNACIONAL DE HIPERTENSIÓN PULMONAR
Estrategia terapéutica en el tratamiento de la hipertensión arterial pulmonar
Sergio V. Perrone*
* Insuficiencia Cardíaca, Hipertensión Pulmonar y Trasplante Cardíaco. Instituto FLENI. Ciudad de Buenos Aires. República Argentina.
Insuficiencia Cardíaca e Hipertensión Pulmonar. Instituto Argentino de Diagnóstico y Tratamiento (IADT). Ciudad de Buenos Aires. Rep. Argentina.
Trasplante Pulmonar y Cardiopulmonar Sanatorio de la Trinidad Mitre. Ciudad de Buenos Aires. República Argentina.
Insuficiencia Cardíaca, Hipertensión Pulmonar y Trasplante Intratorácico. Hospital Italiano de Mendoza. Mendoza. República Argentina.
Correspondencia: Dr. Sergio V. Perrone
Montañeses 2325. CP: 1428. Ciudad de Buenos Aires. República Argentina.
E-mail: svperrone@intermedia.com.ar
Tel.: (54-11) 5777-3200 - Fax: (54-11) 5777-3209
Recibido: 10/12/2008
Aceptado: 10/03/2009
Palabras clave: Hipertensión arterial pulmonar; Estrategia terapéutica; Prostanoides; Oxido nítrico; Bloqueantes de los canales de calcio; Anticoagulación; Antagonistas de la endotelina; Septostomia atrial; Trasplante cardiopulmonar; Hipertensión porto-pulmonar
Introducción
La estrategia y la táctica (Tabla 1) utilizadas para lograr una terapéutica eficaz en el tratamiento de la hipertensión pulmonar tienen un objetivo claro: mejorar los síntomas, la tolerancia al ejercicio, el pronóstico, la calidad de vida y la sobrevida de los pacientes portadores de presión pulmonar elevada (Tabla 2).


Hasta mediados de los años ’80, contábamos con escasas medidas terapéuticas para el tratamiento de la hipertensión arterial pulmonar (HAP). Incluso muy pocos médicos seguían y trataban este tipo de patologías, dado los escasos recursos diagnósticos y terapéuticos existentes que obligaban a catalogar a la mayoría de los pacientes portadores de HAP dentro de la categoría de idiopáticas o de etiología no aclarada y portadores de un muy mal pronóstico a corto plazo.
Para aquellas épocas, nos veíamos obligados a utilizar algunas tácticas y estrategias terapéuticas no convencionales con la finalidad de prolongar la sobrevida o mejorar la calidad de vida de nuestros pacientes. Afortunadamente, en la actualidad disponemos de un número mayor de opciones terapéuticas y se avizora un futuro aun más promisorio.
En este trabajo, nos referiremos al tratamiento médico para el manejo de la HAP, el cual cuenta, en la actualidad, con drogas específicas, drogas no específicas y algunas recomendaciones de índole general que hacen al logro de nuestros objetivos terapéuticos fundamentales (Tabla 2).
Drogas no específicas
Digitálicos
Entre las drogas no específicas, encontramos a la digital, usada desde hace muchos años, sobre todo en aquellos pacientes con signos y síntomas de falla ventricular derecha. La disminución de la función sistólica ventricular derecha es uno de los puntos fundamentales en la progresión de los signos y síntomas de la HAP. La digital, a pesar de poseer un efecto inotrópico positivo débil, parece poseer cierta utilidad en los portadores de HAP con deterioro de la función ventricular derecha1. Si bien no hay estudios que avalen su utilización, suele ser administrada en pacientes con HAP que presentan fibrilación o aleteo auricular con la intención de reducir la frecuencia cardíaca.
Diuréticos
Diuréticos de asa
Los diuréticos de asa son utilizados en aquellos pacientes en los que predominan los signos y síntomas de congestión. Los mismos deben ser utilizados con cuidado, ya que su sobreutilización o abuso provoca una sobreactivación del sistema neurohumoral que resulta perjudicial, pudiendo incrementar la morbimortalidad de este tipo de pacientes y deteriorar la función renal2. Su utilización se encuentra restringida a la reducción de los signos y síntomas de congestión (evitando la brusca reducción de la precarga con la consecuente caída del volumen minuto cardíaco y el deterioro de la función renal).
Antialdosterónicos
Los agentes antialdosterónicos parecen tener una mayor utilidad en el tratamiento de pacientes con HAP y signos de congestión secundaria a falla cardíaca derecha. Sus efectos diuréticos y moduladores del sistema neurohumoral pueden acompañarse de mejorías en el remodelamiento ventricular y del lecho vascular pulmonar3 y, probablemente, deban ser utilizados desde las etapas tempranas del diagnóstico de la hipertensión pulmonar.
Como comentara anteriormente, la utilización de diuréticos debe realizarse con sumo cuidado, la brusca caída de la precarga ventricular puede acompañarse de caída del volumen minuto cardíaco y deterioro de la función renal con incremento de los riesgos para nuestro paciente. Muchas veces, en etapas avanzadas de la patología, debe permitirse cierto grado de congestión, llevando un adecuado control de que la misma no conlleve un deterioro progresivo de la función hepática.
Oxigenoterapia continua domiciliaria
Estos pacientes presentan frecuentemente hipoxemia, y está generalmente admitido por los expertos que cuando su paciente presenta una hipoxemia (PO2 ≤ 60 mm Hg) debería recibir una terapia crónica domiciliaria de oxígeno4-6. Esa terapia puede ser titulada con un test de caminata de 6 minutos, tratando de mantener una saturación arterial de oxígeno superior al 90%.
Drogas específicas
Las drogas específicas actúan sobre los mecanismos fisiopatogénicos.
Anticoagulación
Algunos autores colocan la anticoagulación dentro de la medicación no específica de la HAP. El hecho de ubicar a estas drogas en uno u otro grupo terapéutico (específica o no específica) radica fundamentalmente en la convicción de la intervención de los coágulos y/o trombos, embolizados o in situ, en la génesis y perpetuación de la HAP. La medicación anticoagulante oral fundamenta su utilización en trabajos realizados a principio de los años 80 por Valentín Fuster y colaboradores7, en los cuales la indicación de anticoagulación oral en pacientes con HAP que mantenían un INR entre 1,5 y 2,5 presentaban una mejoría en la clase funcional y en la supervivencia. Si bien este estudio avalaba la utilización de la anticoagulación oral en pacientes portadores de HAP tromboembólica e idiopática, su utilización se ha hecho extensiva (aunque discutida) a la mayoría de los pacientes con HAP debido al elevado hallazgo de trombosis in situ en la HAP de otras causas8-12.
Un apartado especial merece la utilización de la anticoagulación oral en pacientes con hipertensión porto-pulmonar portadores de varices esofágicas, en el cual el riesgo de sangrado es elevado.
Bloqueantes de los canales de calcio
Si bien los bloqueantes de los canales del calcio (BCC) son también ubicados por algunos autores dentro del grupo de terapéuticas no específicas, su efecto beneficioso en algunos casos hacen considerarlo dentro de las terapéuticas que, en determinados momentos de la evolución de la patología pudieran intervenir sobre algunos de los mecanismos fisiopatogénicos de la enfermedad.
Su administración en forma crónica y en dosis elevadas suele mejorar la calidad de vida y la supervivencia de un pequeño grupo de pacientes (< 10%) portadores de HAP que han presentado un test de vasorreactividad positivo13,14.
Los BCC más utilizados son la nifedipina y el diltiazem en dosis que suelen alcanzar 260 mg y 720 mg, respectivamente. Su eficacia suele verse a los pocos meses de tratamiento, pero debe prestarse especial atención al efecto inotrópico negativo de estas drogas que pueden agravar o precipitar la falla ventricular derecha.
En aquellos pacientes que presentan baja frecuencia cardíaca, pueden observarse trastornos de la conducción con la utilización de diltiazem, en cuyo caso está indicada la utilización de una dihidropiridina (nifedipina).
Reguladores del tono vascular
Los reguladores del tono vascular pueden diferenciarse entre: los que actúan sobre los receptores de la endotelina (ET)15,16, los que actúan sobre el óxido nítrico (ON)17,18 y los que actúan sobre las prostaciclinas (prostanoides)18 (Tabla 3).

Prostanoides (Tabla 4)

Los prostanoides son potentes vasodilatadores de corta duración, con efecto también antimitogénico e inhibidor de la agregación plaquetaria. Existen varios prostanoides en uso en la actualidad.
Epoprostenol
El epoprostenol es un prostanoide que se utiliza en infusión continua a través de un catéter central, con una bomba de infusión que permite su administración intravenosa. La experiencia clínica obtenida con esta medicación es interesante y amplia en pacientes con severo deterioro de la capacidad funcional19,20.
Existen algunos problemas relacionados con el catéter central (infecciones) y también, debido a la corta vida media del epoprostenol, si se demora el cambio de la bomba de infusión al recargarla, puede producirse un efecto rebote que puede agravar al paciente21. Por estos motivos, el paciente debe ser muy bien instruido en su utilización, y muchas veces, la misma queda restringida a los ámbitos hospitalarios en pacientes que se encuentran en lista de espera para trasplante. Actualmente, se encuentran en permanente desarrollo catéteres para una administración más segura22.
Iloprost
Otro análogo prostanoide de vida media más prolongada (20 a 25 minutos) que puede administrarse por vía inhalatoria23 o intravenosa (IV)24, es el iloprost. Comúnmente utilizado por vía inhalatoria (6 a 9 nebulizaciones por día), requiere de la utilización de nebulizadores especiales que permiten la administración de partículas entre 3 y 5 µm.
El iloprost puede utilizarse en asociación a otras medicaciones vasodilatadoras (inhibidores de la fosfodiesterasa 5) que veremos posteriormente, permitiendo una disminución relativamente confiable del número de nebulizaciones diarias que debe realizar el paciente.
El estudio AIRE25 demostró que la administración inhalatoria de este prostanoide en pacientes portadores de HAP mejoraba el test de caminata de los 6 minutos en un tratamiento a 12 semanas.
Treprostinil
El treprostinil es otro análogo prostanoide de vida media más prolongada (120 a 180 min.)26 que permite una administración más segura. Se administra por vía subcutánea27 (aunque puede ser administrada también por vía intravenosa22 y se encuentra en estudio su administración por vía inhalatoria28).
La terapia con treprostinil ha demostrado mejorar la supervivencia de pacientes con HAP cuando se la compara con la terapia habitual29,30.
La efectividad de la terapia con treprostinil comparado con el epoprostenol IV es similar, observándose una supervivencia similar en ambos grupos terapéuticos19.
Su ventaja sobre el epoprostenol radica fundamentalmente en su mayor vida media, pues brinda mayor seguridad a su administración en el eventual caso de un retraso en el recambio de la bomba de infusión31. Entre los inconvenientes que suele presentar su administración, podemos mencionar el dolor en el sitio de inyección que muchas veces puede provocar la necesidad de un cambio en la forma de administración, aunque actualmente existen medidas que han permitido controlar este inconveniente en la mayoría de los casos.
Beraprost
El beraprost es otro análogo prostanoide de administración oral32-35. Su absorción es rápida y presenta su concentración máxima entre unos 30 y 40 minutos con una vida media también de 30 a 40 minutos. Esta medicación se encuentra solamente aprobada en Japón y Corea. Su mayor inconveniente es la taquifilaxia y su menor potencia vasodilatadora. Además, aparentemente, no produciría remodelamiento vascular33. Trabajos como los realizados por Vizza y colaboradores y Barst y colaboradores realizados con beraprost evidencian el efecto benéfico inicial y la desarrollada taquifilaxia con beraprost, observando a los 12 meses que su efecto es casi nulo sobre la cantidad de metros en el test de caminata de los 6 minutos36,37.
Oxido nítrico
Otra opción terapéutica de actuar sobre los mecanismos fisiopatogénicos se basa en la utilización de donantes de óxido nítrico (ON) entre los que encontramos algunos bastante conocidos, fundamentalmente, los inhibidores de la fosfodiesterasa 5 (PDE5i)38 como el sildenafilo (Tabla 5).

El sildenafilo ha sido bastante útil, últimamente, en el tratamiento complementario de la hipertensión pulmonar39-43. Su asociación con iloprost (sildenafilo aplicado a pacientes medicados con iloprost), demostraron un efecto favorable en la reducción de la presión pulmonar media, las resistencias vasculares pulmonares, prolongando el efecto vasodilatador obtenido con el iloprost. Si bien la administración de iloprost inhalado produjo una reducción más pronunciada de la PAPm (presión arterial pulmonar media) que el sildenafilo, en este estudio; el agregado de sildenafilo produjo un descenso aun mayor de la PAPm y las RVP (resistencia vascular pulmonar), siendo bastante más interesante la asociación de ambas drogas44. Su utilización en pacientes que reciben antirretrovirales debe realizarse con sumo cuidado por sus interacciones con los mismos requiriendo un ajuste de dosis45.
El tadalafilo es otro inhibidor de la fosfodiesterasa-5, cuyos estudios en hipertensión pulmonar aun se encuentran en curso46,47.
En otro trabajo (PACES), investigadores dirigidos por Simonneau demostraron, en un grupo de 267 pacientes tratados previamente con epoprostenol (por un período no menor a 3 meses), que aquellos pacientes que recibieron sildenafilo mejoraban la distancia caminada en el test de caminata de los 6 minutos y presentaban una mayor reducción de las presiones pulmonares48.
Otra forma de incrementar los niveles de ON a nivel endotelial consiste en administrar precursores del mismo ON. Uno de ellos es la L-arginina49,50. El inconveniente radica en que son necesarias altas dosis de L-Arginina administradas por vía oral para tener un buen efecto sobre los niveles endoteliales de ON.
Otras drogas consideradas donantes de ON son las estatinas. Estas drogas aumentan los niveles endoteliales de ON al prevenir su degradación. Estudios realizados en ratas, revelan un efecto favorable en la reducción de la hipertensión pulmonar provocada por hipoxia51 y abre una nueva posibilidad terapéutica para estas drogas que están actualmente siendo analizadas en estudios en humanos portadores de HAP.
En forma similar, los estimuladores de la guanilato ciclasa soluble (sGCS), nueva familia de drogas con efectos sobre el endotelio vascular, están siendo desarrolladas por laboratorios Bayer, y actualmente hay estudios en curso con los sGCS para el tratamiento de la HAP52.
La guanilato ciclasa soluble (sGC) es una enzima que ayuda a incrementar los niveles de cGMP (guanosin monofosfato cíclico), fundamental en el fenómeno de relajación del las células musculares lisas vasculares, incrementando así los efectos del óxido nítrico53.
Antagonistas de la endotelina (Tabla 6)

Los antagonistas de la endotelina actúan bloqueando la acción de ese potente vasoconstrictor y estimulante de la mitosis que es la endotelina 1 (ET-1) (Figura 1).
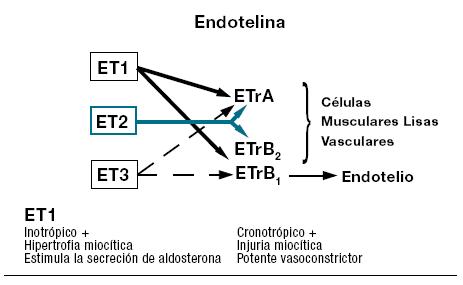
Figura 1. Localizacion de los receptores de endotelina y efectos de la estimulacion.
ET1: Endotelina 1. ET2: Endotelina 2. ET3: Endotelina 3. ETrA: Receptor A de la Endotelina. ETrB1: Recepor B1 de la Endotelina. ETrB2: Receptor B2 de la Endotelina.
Su utilización más intensa en el tratamiento de la HAP surgió luego de que fuera abandonada su intención de utilizarlos en pacientes con insuficiencia cardíaca, debido a alteraciones observadas en los test de función hepática.
A partir de allí, investigadores encontraron que la utilización de este bloqueante no selectivo de los receptores de la ET-1 beneficiaba a los pacientes portadores de HAP a las 16 semanas de tratamiento, observando una mejoría en el test de caminata de los 6 minutos de 44 metros, mejoraba la disnea (índice de Borg), la clase funcional y prolongaba el tiempo al empeoramiento clínico54.
Existen actualmente en el mercado varios antagonistas de la ET-1:
El tezosentan, que actúa en forma no selectiva, antagonizando los efectos de la ET-1 a nivel de los receptores ETA y ETB. Su administración por vía intravenosa ha demostrado reducir las presiones pulmonares55,56.
El bosentan, que actúa en forma competitiva con la ET1 a nivel de los receptores de endotelina-A (ETA) y endotelina- B (ETB)57-59, es efectivo en la reducción de las presiones pulmonares, y al igual que los otros antagonistas de la ET puede producir elevación de enzimas hepáticas reversible en el 10% de los pacientes, aproximadamente54,60,61. Su toxicidad hepática puede verse agravada por su interacción con los anticonceptivos hormonales o la glibenclamida entre otras drogas, lo cual hace necesario un estrecho seguimiento de la función hepática. Posee además efectos teratogénicos y oligospermia.
El sitaxentan, un antagonista selectivo a nivel de los receptores ETA, 6000 más potente que sobre ETB62, ha demostrado también beneficios en la mejoría de la clase funcional de pacientes con HAP tratados por más de 2 años63.
El ambrisentan, un antagonista selectivo a nivel de los receptores ETA64,65 que también ha demostrado mejorías clínicas en pacientes con hipertensión pulmonar, pero que no ha podido desligarse de los efectos colaterales de esta familia de drogas66.
Se debe recordar el frecuente control de la función hepática en los pacientes que reciben este tipo de bloqueantes de los receptores de la ET-1.
Como hemos visto y veremos en los próximos párrafos, varios estudios avalan en la actualidad la terapia simple o combinada con antagonistas de la ET-1.
Terapias combinadas
Actualmente, en muchas oportunidades, ante el fracaso de estas terapéuticas individuales, nos vemos obligados a utilizar combinaciones de drogas cuya asociación se encuentra aun en estudio67-72.
No se dispone actualmente de información adecuada respecto a dosis, interacciones, efectos colaterales, etc. de estas asociaciones, y varios estudios se encuentran en curso, analizando este tipo de combinaciones terapéuticas luego de pequeñas experiencias realizadas con combinaciones como: sildenafilo + bosentan73; sildenafilo + bosentan + iloprost70; bosentan + iloprost + sildenafilo74; iloprost (inh) + bosentan75-77; iloprost (IV) + bosentan77,78 o beraprost + bosentan77 entre otras.
El estudio STEP (bosentan + iloprost vs bosentan + placebo) demostró una significativa mejoría en la clase funcional y en la capacidad de ejercicio de los pacientes que recibieron la asociacion76; en cambio, el estudio COMBI (bosentan + iloprost vs bosentan + placebo), con un número ligeramente inferior de pacientes, fue terminado precozmente por falta de eficacia79.
El estudio BREATH2 (epoprostenol + bosentan vs epoprostenol + placebo) demostró una tendencia a la mejoría clínica y hemodinámica de los pacientes incluidos en la combinación de drogas80.
La combinación de sildenafilo + epoprostenol fue testeada por Simonneau y demostró que en algunos pacientes el agregado de un PDE5i a la medicación con epoprostenol IV puede mejorar la capacidad de ejercicio, las presiones pulmonares y la calidad de vida, demorando los tiempos del empeoramiento clínico48.
Si bien hay estudios que evidenciaron que la administración de bosentan produce una reducción de los niveles plasmáticos de sildenafilo81, han aparecido otros que, basados en la buena tolerancia de la asociación de estas dos drogas, demostraron además mejoría clínica, reducción de las presiones y resistencias pulmonares, incrementando la tolerancia al ejercicio y mejorando las clases funcionales de los pacientes con HAP69,73,82-86.
Iloprost + bosentan87, treprostinil inhalado + bosentan88, también, han demostrado ser bien tolerados y útiles en mejorar los síntomas, la capacidad de ejercicio y la hemodinamia pulmonar de pacientes con HAP. Incluso en pacientes severamente enfermos con insuficiencia hepática e hipertensión porto-pulmonar, la utilización como puente al trasplante hepático de la asociación de prostaciclinas, sildenafilo y bosentan permitió la realización exitosa del mismo70.
Recomendaciones generales
Existen recomendaciones de índole general, la mayoría de ellas aplicables a pacientes portadores de patologías crónicas que deben ser tomadas en consideración en este tipo de pacientes con un objetivo claro, disminuir el impacto negativo de la enfermedad (ansiedad - depresión), evitar situaciones agravantes (tabaco - hipoxia - abuso de drogas - embarazo), prevenir y tratar co-morbilidades. Entre ellas se encuentran: el aporte de un soporte psicológico especializado en el manejo de patologías crónicas avanzadas, un adecuado control del peso corporal con instrucción especializada por un nutricionista, la suspensión de las adicciones (tabaco, alcohol, a otras drogas), el control de la adhesión a las indicaciones médicas (hecho fundamental en el tratamiento de todo paciente crónico), la prevención de patologías infecciosas mediante la educación y la implementación de un esquema completo de vacunación y sobre todo, la profilaxis de la trombosis venosa profunda y de los fenómenos embólicos.
Tratamientos invasivos
Para el manejo de la HAP, se cuenta también con tratamientos invasivos como la septostomía atrial, el trasplante pulmonar o el cardiopulmonar.
Septostomía atrial
Basados en el mejor pronóstico de las cardiopatías congénitas cianóticas con hipertensión pulmonar, surgió la idea de crear un cortocircuito derecha-izquierda a través del foramen ovale. Este procedimiento descomprime las cavidades derechas, mejorando la precarga ventricular izquierda y, consecuentemente, el volumen minuto cardíaco y el transporte tisular de oxígeno, mejorando la clase funcional y reduciendo los niveles de BNP89-91.
Si bien este procedimiento permite una mejoría de la clase funcional de los pacientes, independientemente de una caída de la saturación de oxígeno (PaO2), es utilizado actualmente como un puente hacia el trasplante92,93. El procedimiento debe ser realizado por personal experimentado (mortalidad 5 a 15%) y es aconsejable la realización del cortocircuito en forma lentamente progresiva para evitar una brusca caída de la PaO2, la cual puede incrementar la morbimortalidad del procedimiento94.
Está indicado en pacientes seleccionados, refractarios al tratamiento médico, en clase funcional III-IV de la NYHA (New York Heart Association), con síncope recurrente, insuficiencia cardíaca derecha o como tratamiento paliativo95,96. No se aconseja su realización en pacientes con saturación de O2 por debajo del 90% y en aquellos con hemoglobina menor a 12 gr/dl dado el alto riesgo del procedimiento en este grupo de pacientes94.
Trasplante pulmonar o cardiopulmonar
En casos avanzados, refractarios al tratamiento médico, el trasplante pulmonar y el cardiopulmonar ofrecen una opción terapéutica aceptable, mejorando la hemodinamia pulmonar, la oxigenación y la insuficiencia cardíaca derecha y, consecuentemente, la calidad de vida y la supervivencia94,97.
El procedimiento de preferencia es el trasplante pulmonar bilateral que ofrece cifras de supervivencia a los cinco años cercanas al 50%98. En el caso de que la hipertensión pulmonar se acompañe de un importante deterioro de la función cardíaca, es aconsejable la realización de un trasplante cardiopulmonar en bloque, cuya supervivencia es similar; pero la posibilidad de conseguir bloques cardiopulmonares es significativamente menor que la de conseguir donantes de pulmón solamente.
La escasa cantidad de donantes, lo invasivo del procedimiento y los riesgos quirúrgicos y post quirúrgicos hacen de ésta la última opción terapéutica al igual que en otros tipos de trasplante.
Procedimientos como la administración de epoprostenol intravenoso continuo o la septostomía atrial son frecuentemente utilizados como último puente para la realización del trasplante99. En ciertas patologías como la hemangiomatosis capilar pulmonar (aunque se ha observado recidiva100) o la enfermedad venooclusiva pulmonar, el trasplante pulmonar representa la primera opción terapéutica al no disponer de otras opciones terapéuticas101.
Flujograma terapéutico de la hipertensión pulmonar
Para resumir, según el flujograma terapéutico resuelto en la reunión de Dana Point102,103, además de las medidas generales, se deberán tomar, en primer lugar, las medicaciones basales mediante la realización de un test de vasorreactividad. En base a los resultados del test de vasorreactividad pulmonar, entre los pacientes vasorreactivos, encontraremos aquel grupo de pacientes que poseen mejor respuesta a la administración de bloqueantes de los canales del calcio.
En el grupo de los pacientes sin vasorreactividad, se irán sumando las terapéuticas que hemos mencionado hasta llegar a las combinaciones o a las terapéuticas invasivas (Figura 2).

Figura 2. Flujograma de Dana Point. Tratamiento propuesto para la hipertensión arterial pulmonar basado en la evidencia.
HAPI: hipertensión arterial pulmonar idiopática. BCC: bloqueantes de los canales de calcio. ON: óxido nítrico. PDE 5: fosfodiesterasa tipo 5. SC: subcutánea. EV: endovenosa. OMS: Organización Mundial de la Salud. AREndotelina: antagonista del receptor de endotelina
Opciones terapéuticas especiales
Hipertensión pulmonar con enfermedad cardíaca izquierda
La HAP es una complicación frecuente de la falla cardíaca izquierda o a la valvulopatía mitral y/o aórtica y constituye una de las causas más frecuentes de HAP104,105.
La HAP pasiva secundaria a congestión pulmonar por cardiopatía izquierda es fácilmente reconocible en el cateterismo derecho, presentando presión capilar pulmonar de enclavamiento -wedge- (PCPW) elevada, superior a los 16-18 mm Hg con gradiente transpulmonar (GTP) < de 12 mm Hg. La presencia de una PCPW elevada por encima de 16-18 mm Hg con GTP elevado (> 12 mm Hg) es indicativo de HAP reactiva.
La HAP reactiva implica la presencia de un componente dinámico que con el tiempo puede tornar a ésta en una HAP fija como consecuencia de un daño prolongado y progresivo sobre la estructura vascular pulmonar. Si bien el tratamiento de la HAP con enfermedad cardíaca izquierda consiste en la corrección de la patología izquierda que determina la elevación de las presiones pulmonares, muchas veces, es imprescindible evaluar la reactividad o no de las presiones pulmonares para determinar la conducta terapéutica más adecuada a seguir. Para ello, se utiliza el test de vasorreactividad pulmonar y, luego de la corrección del agente determinante, las presiones pueden manejarse con las medicaciones que hemos enumerado anteriormente105,106.
Hipertensión pulmonar asociada a enfermedad respiratoria o hipoxemia
La hipoxia constituye un factor importante en el incremento de las presiones pulmonares107,110, así, otra causa muy frecuente de HAP la constituyen las patologías parenquimatosas pulmonares como la fibrosis intersticial; enfermedad pulmonar obstructiva crónica (EPOC), otras neuropatías intersticiales o el síndrome de apnea/hipopnea.
La presencia de fibrosis pulmonar, sobre todo en pacientes portadores de patologías del colágeno, produce presiones pulmonares que suelen alcanzar valores relevantes (alrededor de 80 mm Hg). El tratamiento de la enfermedad de base, acompañado de agentes vasodilatadores de última generación se encuentra, actualmente, en evaluación para este tipo de patologías.
En los pacientes con EPOC, la HAP suele ser leve (alrededor de 40 mm Hg) la administración de oxigenoterapia continua domiciliaria y el tratamiento de base de la patología suelen ser suficientes para un buen manejo de las presiones pulmonares. Existen reportes sobre la posibilidad de la inhibición de la vasoconstricción hipóxica pulmonar, provocada por drogas como el sildenafilo o las prostaciclinas que podrían agravar el cuadro clínico. Las neumopatías intersticiales comunes suelen cursar con HAP leve y la mayoría de las veces transitoria, mejorando significativamente con el tratamiento de la enfermedad de base.
La presencia de HAP en pacientes portadores de síndromes de apnea/hipopnea grave se encuentra en constante evaluación, pero la indicación de BiPAP (Biphasic Positive Airway Pressure = Presión bifásica positiva en la vía aérea) en estos pacientes portadores de HAP es indiscutible.
En resumen, la corrección de la hipoxia parece ser una terapéutica altamente eficaz en el tratamiento de estos pacientes que permite la reducción de las presiones pulmonares6.
Cardiopatías congénitas
Otra de las patologías cardiovasculares que cursan frecuentemente con hipertensión pulmonar son algunas de las cardiopatías congénitas111.
Cuando las cardiopatías congénitas incrementan la resistencia vascular pulmonar, se produce una inversión del flujo a través de la comunicación de los circuitos derecho e izquierdo, recibiendo el nombre de síndrome de Eisenmenger. Como comentáramos anteriormente, la observación clínica de una mejor evolución de los pacientes portadores de HAP por síndrome de Eisenmenger que aquellos con HAP idiopática o asociada a otras patologías constituyó uno de los fundamentos para el tratamiento de la HAP avanzada con septostomía atrial progresiva112,113.
Es recomendado el tratamiento anticoagulante (excepto en aquellos pacientes con elevado riesgo de hemorragia), sangrías en aquellos pacientes con marcada poliglobulia, oxigenoterapia ambulatoria permanente, siendo la experiencia con fármacos reguladores del tono vascular relativamente reciente pero con resultados promisorios114.
Hipertensión pulmonar debida a enfermedad tromboembólica crónica
Si bien la presencia de hipertensión pulmonar tromboembólica crónica debe ser sistemáticamente evaluada en pacientes portadores de HAP, su incidencia es baja. El tratamiento anticoagulante de estos pacientes está indicado para mantener un INR más elevado que en otros pacientes portadores de HAP (2,5 a 3) y la tromboendarterectomía pulmonar constituye el tratamiento de elección cuando estos trombos y coágulos son accesibles quirúrgicamente (arterias pulmonares principales, lobares o segmentarias)115,116.
En aquellos pacientes en los cuales el tratamiento quirúrgico no es posible o no está indicado, las opciones terapéuticas habituales son necesarias117. Aunque existe cierta preferencia en la utilización de prostanoides sobre otros reguladores del tono vascular, su beneficio radica en su particular actividad sobre la agregación plaquetaria; últimamente, la intervención sobre los receptores de la endotelina parece estar surtiendo también un efecto beneficioso118,119.
Hipertensión porto-pulmonar
La HAP suele ser una complicación que acompaña frecuentemente a la patología hepática avanzada, contraindicando muchas veces la posibilidad de realizar un trasplante hepático120,121.
La asociación de bloqueantes de los receptores beta y la ligadura o banding de las varices pulmonares pueden prevenir el sangrado de las mismas.
Si bien episodios de sangrado e hipercoagulabilidad se han descripto en patologías hepáticas avanzadas, en estos pacientes no se recomienda la administración rutinaria de anticoagulantes debido al alto riesgo de sangrado122, al igual que la administración de antagonistas de los receptores de la endotelina debido a su toxicidad hepática.
Alguna de las medicaciones enunciadas para el tratamiento de la HAP han sido utilizadas con éxito en el manejo de la HAP de pacientes con hipertensión porto-pulmonar123.
Opciones terapéuticas futuras
Existen algunas drogas en investigación que prometen un futuro aun más interesante en el tratamiento de la HAP. Entre ellas encontramos a los inhibidores de la tyrosin kinasa (TKi) -imatinib-, que ya han sido utilizados en algunos pacientes124.
La progresión de la HAP se encuentra asociada a la migración y proliferación de las células vasculares musculares lisas, proceso el cual se ha vinculado estrechamente con el factor derivado del crecimiento plaquetario (PDGF)125-127, el cual puede ser controlado mediante la administración de TKi124,128. Una terapia dirigida al remodelamiento de la vasculatura pulmonar mediante TKi, como el imatinib, representaría un importante avance en el tratamiento de la patología pulmonar vascular, aunque ciertos alertas sobre la posibilidad de toxicidad cardíaca reportada en su utilización en oncología y su mínima experiencia en humanos reportada en casos de HAP, obligan a más estudios.
Recientemente, en estudios experimentales realizados con un múltiple inhibidor de las kinasas (serina y treonina) llamado sorafenib, que actúa sobre los mecanismos de acción y las causas que generan la HAP, se ha observado prevención del remodelamiento vascular pulmonar mejorando la función pulmonar y cardíaca en ratas129.
El sorafenib ejerce un efecto directo antihipertrófico sobre el miocardio que parece estar mediado a través de la inhibición de la vía de la rafkinasa y su efecto sobre el remodelamiento vascular arterial pulmonar podría brindar beneficios en estos pacientes.
Por otro lado, existen alternativas en la producción del óxido nítrico, que pueden ser mejoradas mediante otras terapéuticas. Es sabido que no todos los pacientes responden de la misma forma a los PDE5i (sildenafilo), posiblemente, debido a alteraciones en el eje de señales ONsGC-cGMP. Existen evidencias derivadas de modelos experimentales que los activadores y estimuladores de la sGC, al puentear la inducción primaria del sistema producida por el ON tendrían un efecto más predecible en la vasodilatación del territorio pulmonar.
Otro sitio sobre el cual es posible actuar, y cuya posibilidad terapéutica se encuentra evaluándose, es la que ofrecen los inhibidores de los transportadores de serotonina130, como el escitalopram. Estos inhibidores específicos de los transportadores de la serotonina como es la 5-hidroxitriptanina (5-HTT) intervienen sobre otro de los mecanismos fisiopatológicos de la HAP y podrían brindar una nueva opción terapéutica131.
Conclusiones
De un pasado ausente de medidas diagnósticas suficientes y terapéuticas efectivas en el tratamiento de la hipertensión pulmonar, en el cual era extremadamente dificultoso encontrar estudios clínicos que evaluaran las posibles terapéuticas a utilizar; la evolución ha sido impactante (Tabla 7), sobre todo en los últimos años en que hemos entrado en una etapa de interés creciente no sólo en la terapéutica, sino en la detección, la evaluación y el tratamiento precoz de esta patología132,133.

El futuro promisorio seguramente nos dará aun más armamentos útiles para combatir esta patología, mejorando su pronóstico y la calidad de vida de los pacientes.
1. Rich S, Seidlitz M, Dodin E, et al. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest 1998;114:787-92. [ Links ]
2. Eshaghian S, Horwich TB, Fonarow GC. Relation of loop diuretic dose to mortality in advanced heart failure. Am J Cardiol 2006;97:1759-64. [ Links ]
3. Nazzareno Galié AM, Massimiliano Palazzini, Luca Negro, Serena Romanazzi and Angelo Branzi. Pharmacological impact on right ventricular remodelling in pulmonary arterial hypertension. European Heart Journal 2007;Supplements (2007) H68-H74. [ Links ]
4. Rhodius E, Caneva J, Sivori M. Argentine Consensus of home long term oxygen therapy. Medicina (B Aires) 1998;58:85-94. [ Links ]
5. Caneva JO, Rabec CA, De Salvo MC, Mazzei JA. Physiopathology, diagnosis and treatment of severe chronic hypoxemia. Role of residential chronic oxygen therapy. Medicina (B Aires) 2001;61:453-69. [ Links ]
6. Ioli F, Braghiroli A, Donner CF. Long-term oxygen therapy. Monaldi Arch Chest Dis 1994;49:9-12. [ Links ]
7. Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984;70:580-7. [ Links ]
8. Johnson SR, Mehta S, Granton JT. Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review. Eur Respir J 2006;28:999-1004. [ Links ]
9. Perel C, Casey M. Anticoagulación en la hipertensión pulmonar. Rev Insuf Cardíaca 2008;3:125-8. [ Links ]
10. Grunig E, Ehlken N, Nagel C. Anticoagulation in pulmonary arterial hypertension. Hamostaseologie 2008;28:225-30. [ Links ]
11. Alam S, Palevsky HI. Standard therapies for pulmonary arterial hypertension. Clin Chest Med 2007;28:91-115, viii. [ Links ]
12. Johnson SR, Granton JT, Mehta S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest 2006;130:545-52. [ Links ]
13. Rich S, Kaufmann E, Levy PS. The effect of high doses of calciumchannel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992;327:76-81. [ Links ]
14. Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005;111:3105-11. [ Links ]
15. Spieker LE, Noll G, Luscher TF. Therapeutic potential for endothelin receptor antagonists in cardiovascular disorders. Am J Cardiovasc Drugs 2001;1:293-303. [ Links ]
16. Luscher TF, Barton M. Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 2000;102:2434-40. [ Links ]
17. Chen HI, Hu CT, Wu CY, Wang D. Nitric Oxide in Systemic and Pulmonary Hypertension. J Biomed Sci 1997;4:244-8. [ Links ]
18. Hankins SR, Horn EM. Current management of patients with pulmonary hypertension and right ventricular insufficiency. Curr Cardiol Rep 2000;2:244-51. [ Links ]
19. McLaughlin VV, Shillington A, Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002;106:1477-82. [ Links ]
20. Sakuma M, Demachi J, Nawata J, Suzuki J, Takahashi T, Shirato K. Long-term epoprostenol therapy in pulmonary artery hypertension. Circ J 2009;73:523-9. [ Links ]
21. Miyaji K, Matsubara H. Continuous intravenous prostacyclin therapy. Nippon Rinsho 2008;66:2139-44. [ Links ]
22. Morris M, Phares K, Zaccardelli D, Ujhelyi MR. A novel catheter system for totally implantable intravenous drug therapy: assessment of catheter function and patency with trepostinil therapy. J Vasc Access 2008;9:20-7. [ Links ]
23. Gessler T, Seeger W, Schmehl T. Inhaled Prostanoids in the Therapy of Pulmonary Hypertension. J Aerosol Med 2008. [ Links ]
24. Hoeper MM, Gall H, Seyfarth HJ, et al. Long-term outcome with intravenous iloprost in pulmonary arterial hypertension. Eur Respir J 2009. [ Links ]
25. Olschewski H, Simonneau G, Galie N, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002;347:322-9. [ Links ]
26. Chattaraj SC. Treprostinil sodium Pharmacia. Curr Opin Investig Drugs 2002;3:582-6. [ Links ]
27. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med 2002;165:800-4. [ Links ]
28. Voswinckel R, Reichenberger F, Gall H, et al. Metered dose inhaler delivery of treprostinil for the treatment of pulmonary hypertension. Pulm Pharmacol Ther 2009;22:50-6. [ Links ]
29. Lang I, Gomez-Sanchez M, Kneussl M, et al. Efficacy of long-term subcutaneous treprostinil sodium therapy in pulmonary hypertension. Chest 2006;129:1636-43. [ Links ]
30. Gómez Sánchez MA. Treprostinil en el tratamiento de la hipertensión arterial pulmonar. Rev Insuf Cardíaca 2008;3:102-4. [ Links ]
31. Vachiery JL, Hill N, Zwicke D, Barst R, Blackburn S, Naeije R. Transitioning from i.v. epoprostenol to subcutaneous treprostinil in pulmonary arterial hypertension. Chest 2002;121:1561-5. [ Links ]
32. Yuki H, Sato S, Arisaka Y, Kato S, Tomoike H. Orally administered beraprost sodium inhibits pulmonary hypertension induced by monocrotaline in rats. Tohoku J Exp Med 1994;173:371-5. [ Links ]
33. Rich J, Hoeper MM. The search for an oral prostanoid to treat pulmonary arterial hypertension continues. Are we getting any closer? Int J Clin Pract Suppl 2009:17-8. [ Links ]
34. Saji T, Ozawa Y, Nakayama T, et al. Short- and long-term effects of the new oral prostacyclin analogue, beraprost sodium, in patients with severe pulmonary hypertension. J Cardiol 1996;27:197-205. [ Links ]
35. Nagaya N, Uematsu M, Okano Y, et al. Effect of orally active prostacyclin analogue on survival of outpatients with primary pulmonary hypertension. J Am Coll Cardiol 1999;34:1188-92. [ Links ]
36. Barst RJ, McGoon M, McLaughlin V, et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003;41:2119-25. [ Links ]
37. Vizza CD, Sciomer S, Morelli S, et al. Long term treatment of pulmonary arterial hypertension with beraprost, an oral prostacyclin analogue. Heart 2001;86:661-5. [ Links ]
38. Eros D, Szantai-Kis C, Kiss R, et al. Structure -activity relationships of PDE5 inhibitors. Curr Med Chem 2008;15:1570-85. [ Links ]
39. Abrams D, Schulze-Neick I, Magee AG. Sildenafil as a selective pulmonary vasodilator in childhood primary pulmonary hypertension. Heart 2000;84:E4. [ Links ]
40. Prasad S, Wilkinson J, Gatzoulis MA. Sildenafil in primary pulmonary hypertension. N Engl J Med 2000;343:1342. [ Links ]
41. Echazarreta D, Mancini L, Curró M, et al. Administración crónica de sildenafil en la hipertensión pulmonar. Rev Insuficiencia Cardíaca 2006;1:23-7. [ Links ]
42. Echazarreta DF, Mancini LF, Curró MF, et al. Tratamiento crónico con inhibidores de la PDE-5 en pacientes portadores de insuficiencia cardíaca avanzada. Rev Insuf Cardíaca 2008;III:16-20. [ Links ]
43. Lema LR. Experiencia clínica con inhibidores de la PDE-5 en hipertensión pulmonar. Rev Insuf Cardíaca 2007;2:163-7. [ Links ]
44. Wilkens H, Guth A, Konig J, et al. Effect of inhaled iloprost plus oral sildenafil in patients with primary pulmonary hypertension. Circulation 2001;104:1218-22. [ Links ]
45. Maur R, Bortman G. Hipertensión pulmonar asociada a infección por el virius de la inmunodeficiencia humana. Rev Insuficiencia Cardíaca 2007;2:41-2. [ Links ]
46. Faruqi S, Fathi H, Morice AH. Combination of sitaxentan and tadalafil for idiopathic pulmonary arterial hypertension following relapse on bosentan. Int J Cardiol 2009. [ Links ]
47. Tay EL, Geok-Mui MK, Poh-Hoon MC, Yip J. Sustained benefit of tadalafil in patients with pulmonary arterial hypertension with prior response to sildenafil: a case series of 12 patients. Int J Cardiol 2008;125:416-7. [ Links ]
48. Simonneau G, Rubin LJ, Galie N, et al. Addition of sildenafil to longterm intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann Intern Med 2008;149:521-30. [ Links ]
49. Nagaya N, Uematsu M, Oya H, et al. Short-term oral administration of L-arginine improves hemodynamics and exercise capacity in patients with precapillary pulmonary hypertension. Am J Respir Crit Care Med 2001;163:887-91. [ Links ]
50. Mehta S, Stewart DJ, Langleben D, Levy RD. Short-term pulmonary vasodilation with L-arginine in pulmonary hypertension. Circulation 1995;92:1539-45. [ Links ]
51. Nishimura T, Vaszar LT, Faul JL, et al. Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation 2003;108:1640-5. [ Links ]
52. Stasch JP, Hobbs AJ. NO-independent, haem-dependent soluble guanylate cyclase stimulators. Handb Exp Pharmacol 2009:277-308. [ Links ]
53. Mittendorf J, Weigand S, Alonso-Alija C, et al. Discovery of Riociguat (BAY 63-2521): A Potent, Oral Stimulator of Soluble Guanylate Cyclase for the Treatment of Pulmonary Hypertension. ChemMedChem 2009. [ Links ]
54. Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002;346:896-903. [ Links ]
55. O'Connor CM, Gattis WA, Adams KF, Jr., et al. Tezosentan in patients with acute heart failure and acute coronary syndromes: design of the Randomized Intravenous Tezosentan study (RITZ-4). Am Heart J 2002;144:583-8. [ Links ]
56. Fitzgerald RK, Oishi P, Ovadia B, et al. Tezosentan, a combined parenteral endothelin receptor antagonist, produces pulmonary vasodilation in lambs with acute and chronic pulmonary hypertension. Pediatr Crit Care Med 2004;5:571-7. [ Links ]
57. Russell FD, Davenport AP. Characterization of endothelin receptors in the human pulmonary vasculature using bosentan, SB209670, and 97-139. J Cardiovasc Pharmacol 1995;26 Suppl 3:S346-7. [ Links ]
58. Breu V, Ertel SI, Roux S, Clozel M. The pharmacology of bosentan. Expert Opin Investig Drugs 1998;7:1173-92. [ Links ]
59. Clozel M, Roux S. The pharmacology of endothelin and its antagonist bosentan. Ann Endocrinol (Paris) 2000;61:75-9. [ Links ]
60. Channick R, Badesch DB, Tapson VF, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary hypertension: a placebo-controlled study. J Heart Lung Transplant 2001;20:262-3. [ Links ]
61. Dietrich CG, Geier A, Lammert F. Bosentan for pulmonary hypertension. N Engl J Med 2002;347:292-4; author reply -4. [ Links ]
62. MacIntyre IM, Dhaun N, Goddard J, Webb DJ. Sitaxsentan sodium for pulmonary hypertension. Drugs Today (Barc) 2008;44:585-600. [ Links ]
63. Scott LJ. Sitaxentan: in pulmonary arterial hypertension. Drugs 2007;67:761-70; discussion 71-2. [ Links ]
64. Billman GE. Ambrisentan (Myogen). Curr Opin Investig Drugs 2002;3:1483-6. [ Links ]
65. Jacobs A, Preston IR, Gomberg-Maitland M. Endothelin receptor antagonism in pulmonary arterial hypertension--a role for selective ET(A) inhibition? Curr Med Res Opin 2006;22:2567-74. [ Links ]
66. Galie N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2005;46:529-35. [ Links ]
67. Beyer S, Speich R, Fischler M, Maggiorini M, Ulrich S. Long-term experience with oral or inhaled vasodilator combination therapy in patients with pulmonary hypertension. Swiss Med Wkly 2006;136:114-8. [ Links ]
68. Porhownik NR, Al-Sharif H, Bshouty Z. Addition of sildenafil in patients with pulmonary arterial hypertension with inadequate response to bosentan monotherapy. Can Respir J 2008;15:427-30. [ Links ]
69. Burgess G, Hoogkamer H, Collings L, Dingemanse J. Mutual pharmacokinetic interactions between steady-state bosentan and sildenafil. Eur J Clin Pharmacol 2008;64:43-50. [ Links ]
70. Austin MJ, McDougall NI, Wendon JA, et al. Safety and efficacy of combined use of sildenafil, bosentan, and iloprost before and after liver transplantation in severe portopulmonary hypertension. Liver Transpl 2008;14:287-91. [ Links ]
71. Pulido Zamudio T. Pharmacological treatment of pulmonary arterial hypertension. Arch Cardiol Mex 2007;77 Suppl 4:S4-198-201. [ Links ]
72. Naeije R, Huez S. Expert opinion on available options treating pulmonary arterial hypertension. Expert Opin Pharmacother 2007;8:2247-65. [ Links ]
73. Hoeper MM, Faulenbach C, Golpon H, Winkler J, Welte T, Niedermeyer J. Combination therapy with bosentan and sildenafil in idiopathic pulmonary arterial hypertension. Eur Respir J 2004;24:1007-10. [ Links ]
74. Catapano-Minotti G, Corsonello A, Guadalupi G, Spani R, Antonelli-Incalzi R. Treatment of severe pulmonary hypertension secondary to scleroderma: a three-drug approach. Intern Med 2008;47:511-3. [ Links ]
75. Hoeper MM, Taha N, Bekjarova A, Gatzke R, Spiekerkoetter E. Bosentan treatment in patients with primary pulmonary hypertension receiving nonparenteral prostanoids. Eur Respir J 2003;22:330-4. [ Links ]
76. McLaughlin VV, Oudiz RJ, Frost A, et al. Randomized study of adding inhaled iloprost to existing bosentan in pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:1257-63. [ Links ]
77. Seyfarth HJ, Pankau H, Hammerschmidt S, Schauer J, Wirtz H, Winkler J. Bosentan improves exercise tolerance and Tei index in patients with pulmonary hypertension and prostanoid therapy. Chest 2005;128:709-13. [ Links ]
78. Halank M, Kolditz M, Miehlke S, Schiemanck S, Schmeisser A, Hoeffken G. Combination therapy for portopulmonary hypertension with intravenous iloprost and oral bosentan. Wien Med Wochenschr 2005;155:376-80. [ Links ]
79. Hoeper MM, Seyfarth HJ, Hoeffken G, et al. Experience with inhaled iloprost and bosentan in portopulmonary hypertension. Eur Respir J 2007;30:1096-102. [ Links ]
80. Humbert M, Barst RJ, Robbins IM, et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J 2004;24:353-9. [ Links ]
81. Paul GA, Gibbs JS, Boobis AR, Abbas A, Wilkins MR. Bosentan decreases the plasma concentration of sildenafil when coprescribed in pulmonary hypertension. Br J Clin Pharmacol 2005;60:107-12. [ Links ]
82. Minai OA, Arroliga AC. Long-term results after addition of sildenafil in idiopathic PAH patients on bosentan. South Med J 2006;99:880-3. [ Links ]
83. Mathai SC, Girgis RE, Fisher MR, et al. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J 2006. [ Links ]
84. Lunze K, Gilbert N, Mebus S, et al. First experience with an oral combination therapy using bosentan and sildenafil for pulmonary arterial hypertension. Eur J Clin Invest 2006;36 Suppl 3:32-8. [ Links ]
85. Kamata Y, Iwamoto M, Minota S. Consecutive use of sildenafil and bosentan for the treatment of pulmonary arterial hypertension associated with collagen vascular disease: sildenafil as reliever and bosentan as controller. Lupus 2007;16:901-3. [ Links ]
86. Mathai SC, Girgis RE, Fisher MR, et al. Addition of sildenafil to bosentan monotherapy in pulmonary arterial hypertension. Eur Respir J 2007;29:469-75. [ Links ]
87. Halank M, Kolditz M, Opitz C, Hoeffken G, Ewert R. Successful switch from long-term intravenous iloprost to non-invasive combination therapy in idiopathic pulmonary arterial hypertension. Wien Klin Wochenschr 2006;118:54-9. [ Links ]
88. Channick RN, Olschewski H, Seeger W, Staub T, Voswinckel R, Rubin LJ. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial hypertension. J Am Coll Cardiol 2006;48:1433-7. [ Links ]
89. O'Byrne ML, Rosenzweig ES, Barst RJ. The effect of atrial septostomy on the concentration of brain-type natriuretic peptide in patients with idiopathic pulmonary arterial hypertension. Cardiol Young 2007;17:557-9. [ Links ]
90. Nihill MR, O'Laughlin MP, Mullins CE. Effects of atrial septostomy in patients with terminal cor pulmonale due to pulmonary vascular disease. Cathet Cardiovasc Diagn 1991;24:166-72. [ Links ]
91. Kerstein D, Levy PS, Hsu DT, Hordof AJ, Gersony WM, Barst RJ. Blade balloon atrial septostomy in patients with severe primary pulmonary hypertension. Circulation 1995;91:2028-35. [ Links ]
92. Rothman A, Beltran D, Kriett JM, Smith C, Wolf P, Jamieson SW. Graded balloon dilation atrial septostomy as a bridge to lung transplantation in pulmonary hypertension. Am Heart J 1993;125:1763-6. [ Links ]
93. Lagioia AM, Atamañuk N, Bortman G. Tratamniento intervencionista de la hipertensión pulmonar. Rev Insuficiencia Cardíaca 2007;2:137-40. [ Links ]
94. Sager JS, Ahya VN. Surgical therapies for pulmonary arterial hypertension. Clin Chest Med 2007;28:187-202, ix. [ Links ]
95. Sobrino N, Frutos A, Calvo L, Casamayor LM, Arcas R. Palliative interatrial septostomy in severe pulmonary hypertension. Rev Esp Cardiol 1993;46:125-8. [ Links ]
96. Law MA, Grifka RG, Mullins CE, Nihill MR. Atrial septostomy improves survival in select patients with pulmonary hypertension. Am Heart J 2007;153:779-84. [ Links ]
97. Kreider M, Kotloff RM. Selection of candidates for lung transplantation. Proc Am Thorac Soc 2009;6:20-7. [ Links ]
98. Christie JD, Edwards LB, Aurora P, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fifth official adult lung and heart/lung transplantation report--2008. J Heart Lung Transplant 2008;27:957-69. [ Links ]
99. Rothman A, Sklansky MS, Lucas VW, et al. Atrial septostomy as a bridge to lung transplantation in patients with severe pulmonary hypertension. Am J Cardiol 1999;84:682-6. [ Links ]
100.de Perrot M, Waddell TK, Chamberlain D, Hutcheon M, Keshavjee S. De novo pulmonary capillary hemangiomatosis occurring rapidly after bilateral lung transplantation. J Heart Lung Transplant 2003;22:698-700. [ Links ]
101.Montani D, Price LC, Dorfmuller P, et al. Pulmonary veno-occlusive disease. Eur Respir J 2009;33:189-200. [ Links ]
102.Olschewski H. Dana Point: what is new in the diagnosis of pulmonary hypertension?. Dtsch Med Wochenschr 2008;133 Suppl 6:S180-2. [ Links ]
103.Hoeper MM, Ghofrani HA, Grimminger F, Rosenkranz S. Dana Point: what is new in the treatment of pulmonary hypertension?. Dtsch Med Wochenschr 2008;133 Suppl 6:S191-5. [ Links ]
104.Heresi GA, Dweik RA. Pulmonary hypertension: evaluation and management. Compr Ther 2007;33:150-61. [ Links ]
105.Bonderman D, Martischnig AM, Moertl D, Lang IM. Pulmonary hypertension in chronic heart failure. Int J Clin Pract Suppl 2009:4-10. [ Links ]
106.Shah RV, Semigran MJ. Pulmonary hypertension secondary to left ventricular systolic dysfunction: contemporary diagnosis and management. Curr Heart Fail Rep 2008;5:226-32. [ Links ]
107.Dagher IK, Mishalany HG, Simeone FA, Wilson JL. Mechanisms of pulmonary hypertension in acute hypoxia. J Urol Nephrol (Paris) 1961;42:743-54. [ Links ]
108.Dalen JE, Bruce RA, Cobb LA. Interaction of chronic hypoxia of moderate altitude on pulmonary hypertension complicating defect of the atrial septum. N Engl J Med 1962;266:272-7. [ Links ]
109.Reeves JT, Leathers JE, Eiseman B, Spencer FC. Alveolar hypoxia versus hypoxemia in the development of pulmonary hypertension. Med Thorac 1962;19:561-72. [ Links ]
110.Hopkins RA, Wolfe WG. Role of hypoxia in development of pulmonary hypertension. Surg Forum 1978;29:179-81. [ Links ]
111. Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation 2007;115:1039-50. [ Links ]
112.Dimopoulos K, Giannakoulas G, Wort SJ, Gatzoulis MA. Pulmonary arterial hypertension in adults with congenital heart disease: distinct differences from other causes of pulmonary arterial hypertension and management implications. Curr Opin Cardiol 2008;23:545-54. [ Links ]
113.Diller GP, Dimopoulos K, Broberg CS, et al. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: a combined retrospective and case-control study. Eur Heart J 2006;27:1737-42. [ Links ]
114.Galie N, Manes A, Palazzini M, et al. Management of pulmonary arterial hypertension associated with congenital systemic-to-pulmonary shunts and Eisenmenger's syndrome. Drugs 2008;68:1049-66. [ Links ]
115.Porres-Aguilar M, Anaya-Ayala JE, Porres-Munoz M, Bracamontes F. Pulmonary thromboendarterectomy in patients with chronic thromboembolic pulmonary hypertension. Cir Cir 2007;75:131-8. [ Links ]
116.D'Armini AM, Zanotti G, Pozzi M, et al. Surgical treatment of chronic thromboembolic pulmonary hypertension with pulmonary endarterectomy. G Ital Cardiol (Rome) 2006;7:454-63. [ Links ]
117.Porres-Aguilar M, Anaya-Ayala JE, Porres-Munoz M, Bracamontes F. Chronic thromboembolic pulmonary hypertension. Cir Cir 2007;75:123-30. [ Links ]
118.Kaplinsky EJ. Utilidad del bosentan en la hipertensión pulmonar tromboembólica crónica: Estudio BENEFIT Bosentan for inoperable chronic thromboembolic pulmonary hypertension (CTEPH): A randomized, placebo-controlled trial: BENEFIT Jais X, Ghofrani A, Hoeper MM, et al. Am J Respir Crit Care Med 2007; 175:A896 (Abst D82). Rev Insuf Cardíaca 2008;3:63-4. [ Links ]
119.Tomas C, Callejas-Rubio JL, Rios-Fernandez R, Ortego-Centeno N. Bosentan and chronic thromboembolic pulmonary hypertension. Med Clin (Barc) 2007;129:639. [ Links ]
120.Yeshua H, Blendis LM, Oren R. Pulmonary manifestations of liver diseases. Semin Cardiothorac Vasc Anesth 2009;13:60-9. [ Links ]
121.Koch DG, Caplan M, Reuben A. Pulmonary hypertension after liver transplantation: case presentation and review of the literature. Liver Transpl 2009;15:407-12. [ Links ]
122.Northup PG. Hypercoagulation in liver disease. Clin Liver Dis 2009;13:109-16. [ Links ]
123.Sakai T, Planinsic RM, Mathier MA, de Vera ME, Venkataramanan R. Initial experience using continuous intravenous treprostinil to manage pulmonary arterial hypertension in patients with end-stage liver disease. Transpl Int 2009;22:554-61. [ Links ]
124.Frenckner B, Broome M, Lindstrom M, Radell P. Platelet-derived growth factor inhibition--a new treatment of pulmonary hypertension in congenital diaphragmatic hernia? J Pediatr Surg 2008;43:1928-31. [ Links ]
125.Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology (Oxford) 2008;47 Suppl 5:v2-4. [ Links ]
126.Selimovic N, Bergh CH, Andersson B, Sakiniene E, Carlsten H, Rundqvist B. Growth factors and interleukin-6 across the lung circulation in pulmonary hypertension. Eur Respir J 2009. [ Links ]
127.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes development 2008; 22:1276-312. [ Links ]
128.Schermuly R, Grimminger F. Update in basic research in the therapy of pulmonary arterial hypertension. Dtsch Med Wochenschr 2008;133 Suppl 6:S170-2. [ Links ]
129.Klein M, Schermuly RT, Ellinghaus P, et al. Combined tyrosine and serine/ threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation 2008;118:2081-90. [ Links ]
130.Maclean MR, Dempsie Y. Serotonin and pulmonary hypertension-from bench to bedside? Curr Opin Pharmacol 2009. [ Links ]
131.Shah SJ, Gomberg-Maitland M, Thenappan T, Rich S. Selective Serotonin Reuptake Inhibitors and the Incidence and Outcome of Pulmonary Hypertension. Chest 2009. [ Links ]
132.Rhodes CJ, Davidson A, Gibbs JS, Wharton J, Wilkins MR. Therapeutic targets in pulmonary arterial hypertension. Pharmacol Ther 2009;121:69-88. [ Links ]
133.Olsson KM, Hoeper MM. Novel approaches to the pharmacotherapy of pulmonary arterial hypertension. Drug Discov Today 2009;14:284-90. [ Links ]

