










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkInsuficiencia cardíaca
On-line version ISSN 1852-3862
Insuf. card. vol.4 no.3 Ciudad Autónoma de Buenos Aires July/Sept. 2009
ARTÍCULO DE ACTUALIZACIÓN
Muerte súbita en deportistas
Importancia del reconocimiento de las miocardiopatías
América Pérez*#, Jorge González Zuelgaray*
* Instituto Argentino de Diagnóstico y Tratamiento (IADT). Ciudad de Buenos Aires. República Argentina.
# Sanatorio "Dr. Julio Méndez". Ciudad de Buenos Aires. República Argentina.
Correspondencia: Dr. Jorge González Zuelgaray
Marcelo T. de Alvear 2346.
CP: 1122. Ciudad de Buenos Aires. República Argentina.
Fax: (54-11) 4963-9500 Interno #329.
E-mail: jgz1953@gmail.com.
Recibido: 18/08/2009
Aceptado: 10/09/2009
Palabras clave: Insuficiencia cardíaca; Miocardiopatía; Muerte súbita
Introducción
La muerte súbita (MS) cardíaca ha sido definida como aquella "debida a causas cardíacas, de inicio abrupto y rápida evolución, de manera que se produce dentro de la primera hora del inicio de los síntomas agudos, y en la cual, aún en conocimiento de una enfermedad cardíaca preexistente, la manera de presentarse resulta inesperada"1. La incidencia anual de MS estimada en la población general es de 1:10002, y si bien ocurre con mayor frecuencia en la segunda mitad de la vida, cuando el afectado es un individuo joven y todavía más si es deportista, se convierte en un hecho impactante. Aunque se ha estimado que la ocurrencia anual de MS en atletas jóvenes (menores de 35 años) alcanza a 1 caso en 200 mil deportistas, probablemente estas cifras sean menores a las reales3.
En primer término, se debe reconocer que las causas de la MS en los deportistas jóvenes son diferentes a aquellas identificadas para la población general. De acuerdo con los registros de autopsias, en los Estados Unidos la miocardiopatía hipertrófica es la causa más importante (36%), seguida por la anomalía del origen de las arterias coronarias (17%), la miocarditis (6%), la displasia arritmogénica (4%) y por último, con el 4% de los casos, las canalopatías (síndromes de QT prolongado, de Brugada, de QT corto y taquicardia ventricular catecolaminérgica)4. En el presente artículo no se considerarán las canalopatías, debido a que no constituyen el área de interés de la revista "Insuficiencia Cardíaca".
Cardiopatías vinculadas con la MS en deportistas
Miocardiopatía hipertrófica
Se caracteriza por la hipertrofia del ventrículo izquierdo con desarreglo estructural de los miocardiocitos y fibrosis5,6. Se presenta en 1 cada 500 nacimientos7, y hay consenso en plantear dicho diagnóstico cuando el espesor de la pared del ventrículo izquierdo supera los 15 mm sin una causa que lo justifique8. Puede sospecharse entre el 75 y el 95% de los casos mediante un electrocardiograma (ECG) de 12 derivaciones9. Son factores de riesgo para la ocurrencia de MS: el síncope, la historia familiar de MS, el registro de taquicardia ventricular no sostenida en el Holter, la respuesta plana o la disminución de la presión arterial durante el ejercicio, la hipertrofia ventricular severa y la presencia de gradiente en el tracto de salida del ventrículo izquierdo10.
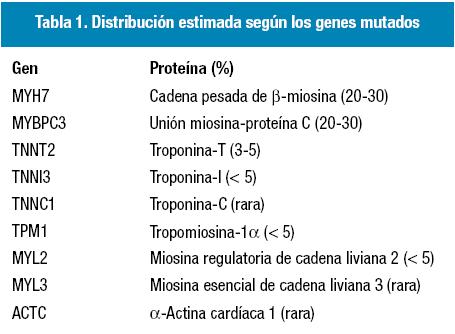
Dado que hasta en el 90% de los casos se trata de una enfermedad familiar, el mayor avance de los últimos años ha provenido de la Genética. Se sabe que el patrón de herencia es autosómico dominante (salvo en los casos con mutaciones en el ADN mitocondrial que se heredan por vía materna) y se han identificado más de 400 mutaciones11. Sin embargo, el 60% de los pacientes con miocardiopatía hipertrófica presenta alteraciones en 9 genes codificantes de proteínas estructurales del sarcómero del músculo cardíaco12-21. En la Tabla 1, se muestra la distribución estimada de los pacientes con miocardiopatía hipertrófica de acuerdo con la identificación de los genes mutados22.
También, se han detectado mutaciones en genes involucrado en el metabolismo energético de las mitocondrias, como el síndrome de hipertrofia ventricular izquierda asociado con preexcitación. El origen de esta entidad se basa en alteraciones de los genes que codifican la subunidad γ2 de la proteinquinasa dependiente del AMP cíclico (PRKAG2)23. Esta entidad morfológicamente idéntica a la miocardiopatía hipertrófica obedece a una alteración en el metabolismo del glucógeno.
Hasta el momento, se considera que las mutaciones no predicen el fenotipo, ya que en una misma familia pueden existir individuos con diferentes grados de hipertrofia o con mayor predisposición a MS que otros con la misma mutación. Esto se debe a la intervención de genes modificadores y polimorfismos que requieren estudios más exhaustivos24,25. Sin embargo, la identificación del genotipo podría contribuir a la estratificación del riesgo, lo que constituye el aspecto relevante para los individuos afectos a las prácticas deportivas.
¿Miocardiopatía hipertrófica o hipertrofia exagerada frente al esfuerzo?
La baja prevalencia de miocardiopatía hipertrófica en autopsias de poblaciones militares jóvenes, en las cuales la MS ocurrió durante la actividad física vigorosa, resulta llamativa26.
La hipertrofia constituye una respuesta adaptativa durante el entrenamiento deportivo, con valores que muchas veces coinciden con los aceptados para el diagnóstico de miocardiopatía hipertrófica27-29. Curiosamente, el notable predominio en varones de la MS que ocurre durante la práctica deportiva no coincide con la distribución de la miocardiopatía hipertrófica, que no muestra diferencias según el género30. En la misma línea, no ha habido reportes de miocardiopatía hipertrófica en autopsias de deportistas menores de 13 años que fallecieron súbitamente, a pesar de que esta entidad se hace manifiesta en la infancia31. Esto sugiere que la hipertrofia observada en varones en edad puberal o mayores podría constituir una respuesta del miocardio sujeto a un estímulo reiterado en presencia de niveles más elevados de testosterona. En este sentido, Koenig y col. observaron mayor hipertrofia ventricular en roedores machos en comparación con las hembras luego de ejercicio crónico, lo que revirtió con la orquidectomía32. El predominio de varones de raza negra fallecidos súbitamente durante prácticas deportivas y con diagnóstico de miocardiopatía hipertrófica en las autopsias debe ser analizado a la luz de la mayor respuesta hipertrófica que muestra la población afroamericana, frente a iguales valores de presión arterial, en comparación con los individuos de raza blanca33.
Por otra parte, la actividad adrenérgica continuada a la que son expuestos los atletas durante períodos prolongados no sólo predispone a arritmias complejas, sino que es un poderoso estímulo para el desarrollo de modificaciones estructurales que podrían simular la miocardiopatía hipertrófica34, con las consiguientes alteraciones electrofisiológicas: prolongación de la refractariedad35, reducción en la amplitud del potencial de acción36, y en el electrocardiograma: bloqueos AV, arritmias ventriculares y alteraciones en la repolarización.
En conclusión, las características demográficas de la MS en deportistas jóvenes con hipertrofia ventricular no coinciden con las de la miocardiopatía hipertrófica y podrían corresponder a una respuesta hipertrófica exagerada frente al estímulo que representa el entrenamiento asociado a una descarga adrenérgica repetida e intensa en presencia de un efecto potenciador de la testosterona. Dado que en individuos predispuestos, la hipertrofia dejaría de ser "fisiológica" para simular una miocardiopatía hipertrófica (lo que no es fácil de distinguir sólo con criterios anatómicos35), el desafío mayor consiste en identificar a aquellos individuos en riesgo de presentar tal respuesta exagerada de modo de prevenir así la ocurrencia de MS37. Con la mayor sensibilidad diagnóstica que van ganando los estudios genéticos, probablemente en algún momento radique en dichas determinaciones la clave para atacar el problema. En la actualidad, se identifica una mutación en el 30-61% de los pacientes con miocardiopatía hipertrófica38, lo que seguramente ha de aumentar e inclusive permitirá una detección "en cascada" de los familiares portadores de la mutación, con el consiguiente efecto beneficioso para reducir la MS, no sólo entre los deportistas, sino también en la población general.
Anomalías coronarias
Si bien entre los deportistas mayores de 40 años que padecen MS, la enfermedad coronaria aterosclerótica constituye el principal hallazgo de la autopsia, por debajo de los 35 años las anomalías coronarias (principalmente, origen anómalo o trayectos intramiocárdicos) constituyen en los Estados Unidos la segunda causa de MS (17%)4.
Lamentablemente, el ECG y la prueba de esfuerzo no son de utilidad en estas situaciones. En cambio, es valioso el interrogatorio en busca de síntomas como síncope o precordialgia que ocurren durante el ejercicio. En esos casos, el ecocardiograma en manos expertas puede aportar una valiosa información y, de no haber un resultado concluyente, tanto las nuevas técnicas (resonancia magnética o tomografía computarizada multicorte) como la cinecoronariografía convencional permiten definir con precisión las características de la circulación coronaria.
Displasia arritmogénica del ventrículo derecho
Se caracteriza por la sustitución progresiva (desde el subepicardio hacia el endocardio) del miocardio por tejido adiposo y por fibrosis39. Si bien se localiza con preferencia en el ventrículo derecho, también se observa en el ventrículo izquierdo40, por lo que habría que denominarla "displasia arritmogénica" o "miocardiopatía arritmogénica".
Se calcula que su prevalencia es de 1 cada 5000 individuos, con predominio en los varones. La mayoría de los casos son diagnosticados antes de los 40 años de edad. Es habitual la presencia de arritmias ventriculares sintomáticas que se originan en el ventrículo derecho y pueden causar síncope y MS. Esta entidad causa el 5% del total de casos de MS en atletas jóvenes41,42, y en Europa (particularmente en Italia) es la primera causa de MS en deportistas43. No resulta claro, si este alto índice de diagnósticos en deportistas se debe a que las arritmias ventriculares son a menudo inducidas por el esfuerzo, o si el ejercicio facilita la aparición de anomalías estructurales precondicionadas genéticamente44.
En relación con la notable diferencia en la frecuencia de observación de displasia entre Europa y los Estados Unidos, creemos que no se debe sólo a diferencias regionales de presentación, sino a un relativo desconocimiento de esta entidad en Norteamérica. Esto ha comenzado a revertirse a partir de la creación de un registro conjunto europeoestadounidense de displasia arritmogénica.
Tradicionalmente, se recurre a la "suma de puntos" -con criterios mayores y menores- para realizar el diagnóstico (Tabla 2)45, lo que muestra las dificultades para asegurar un diagnóstico en vida del paciente.

La etiología y la patogenia de la displasia arritmogénica no son del todo conocidas. Se ha propuesto una anomalía del desarrollo con atrofia miocárdica progresiva, y se le adjudica al estrés mecánico un papel como promotor de apoptosis y degeneración fibroadiposa46.
El origen genético de la enfermedad, con un patrón de herencia autonómico dominante, surge de la descripción de formas familiares47. En su inicio, los estudios genéticos detectaron anomalías en los cromosomas 14 (14q23-q24 y 14q12-q22)48,49 y 1 (1q42-q43)50. Los loci 14q23 y 1q42 contienen genes que codifican la α-actinina, una proteína básica en la unión de la estructura sarcomérica a la pared del miocito.
Hasta la fecha, se llevan identificadas anomalías en 6 genes: 4 codifican proteínas del desmosoma (estructura de unión intercelular), y hasta el 45% de los pacientes presenta una mutación que afecta a la proteína placofilina 2 (PKP2)51-57. Anomalías en proteínas como desmoplaquina, placoglobina, desmogleína-2 o desmocolina-2 aparecen raramente en pacientes con displasia arritmogénica58-60. La participación de tantos loci en las alteraciones genéticas de esta entidad condiciona su clasificación en distintos fenotipos (Tabla 3)61.

Además de las formas aisladas, la displasia también se presenta como parte del síndrome de Naxos (por encontrarse los casos en la isla del mismo nombre en el mar Egeo). Dicho síndrome se caracteriza por displasia arritmogénica, queratodermia palmoplantar y cabello lanudo, y se transmite en forma autosómica recesiva.
La displasia se debe diferenciar de la enfermedad de Uhl, que aparece fundamentalmente en niños y adolescentes. En esta última, no existe un componente familiar, su presentación habitual es la insuficiencia cardíaca y el estudio anatomopatológico muestra una falta completa de miocardio sin depósitos grasos.
Siempre se debe descartar la displasia arritmogénica en pacientes reanimados de un paro cardíaco con sospecha de síndrome de Brugada o de fibrilación ventricular idiopática. La ausencia de cardiopatía estructural que define a estas dos entidades es frecuente que se limite a estudios de imágenes macroscópicas que no alcanzan para las formas incipientes de la displasia. Es de esperar que el diagnóstico genético contribuya a un diagnóstico más preciso.
La estratificación del riesgo de MS en estos pacientes adolece de falta de grandes estudios prospectivos. La información aportada por el seguimiento de pacientes con cardiodesfibriladores implantables se convierte, por el momento, en el elemento más valioso en este sentido. Así, las descargas apropiadas de los dispositivos se vinculan con la ocurrencia de síncope, afectación del ventrículo izquierdo, arritmias ventriculares sintomáticas e historia familiar de MS62,63.
Hacia una efectiva prevención de la MS en deportistas
Está demostrado que los atletas competitivos con cardiopatías tienen un riesgo de MS 3 veces mayor que el de la población no deportista64.
La evaluación electrocardiográfica, si se acompaña de una interpretación de los trazados a cargo de médicos con probada experiencia, permite salvar vidas. Sin embargo, en tanto la Sociedad Europea de Cardiología y el Comité Olímpico Internacional recomiendan incluir el ECG en la evaluación previa a la autorización de prácticas deportivas competitivas, la American Heart Association sólo indica un interrogatorio y examen físico, en gran medida sobre la base de los elevados costos de la medicina estadounidense, que afortunadamente en nuestro país son enormemente menores.
La detección de cardiopatías mediante estudios genéticos no es posible en la actualidad debido a los costos, las demoras y el hallazgo aun limitado de los genes causantes. El estudio genético, en cambio, tiene gran valor y está justificado en individuos con diagnóstico probable, pero no confirmado, de afecciones cardíacas causantes de MS, en especial en presencia de síntomas difíciles de jerarquizar (como síncope o presíncope, que tanto pueden deberse a un trastorno neurocardiogénico como a arritmias malignas).
La educación de la población general y la educación médica continuada constituyen aliados imprescindibles para la prevención de la MS en deportistas. En este sentido, resulta notable la escasa calidad de los datos aportados por los equipos médicos de los diferentes países participantes en el Campeonato Mundial de Fútbol de 200665. Esto resulta trascendente cuando se trata de la interpretación electrocardiográfica, ya que sólo reduciendo al máximo los falsos positivos y negativos es posible optimizar la relación costo-beneficio de los programas de detección a gran escala.
La existencia de un marco regulatorio a nivel nacional resulta viable en países con poblaciones como las de Italia o de la Argentina, en tanto es sumamente difícil en países con enorme población como, por ejemplo, nuestro vecino Brasil. Dichas normas deben incluir la obligatoriedad de la realización de estudios previos a la práctica deportiva, la disponibilidad de desfibriladores externos automáticos en clubes, gimnasios y campos de deportes, la capacitación de los entrenadores en técnicas de reanimación cardiopulmonar y, frente a lo inevitable, la realización de necropsias en todos los casos de MS ocurrida durante o luego de la finalización de prácticas deportivas (lo que contribuiría al conocimiento de la distribución regional de los fenotipos).
En nuestra opinión, se pueden esperar significativos avances en no más de una década si a las medidas adecuadas de prevención se suman los extraordinarios avances que ha de aportar la Biología Molecular.
1. Priori SG, Aliot E, Blomstrom-Lundqvist C, Bossaert L, Breithardt G, Brugada P, Camm AJ, Cappato R, Cobbe SM, Di Mario C, Maron BJ, McKenna WJ, Pedersen AK, Ravens U, Schwartz PJ, Trusz-Gluza M, Vardas P, Wellens HJ, Zipes DP: Task Force on Sudden Cardiac Death of the European Society of Cardiology. Eur Heart J 2001;22:1374-450. [ Links ]
2. Straus SM, Bleumink GS, Dieleman JP, van der Lei J, Stricker BH, Sturkenboom MC: The incidence of sudden cardiac death in the general population. J Clin Epidemiol 2004;57:98-102. [ Links ]
3. Maron BJ, Gohman TE, Aeppli D. Prevalence of sudden cardiac death during competitive sports activities in Minnesota high school athletes. J Am Coll Cardiol 1998;32:1881-4. [ Links ]
4. Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO. Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the United States, 1980-2006. Circulation 2009;119:1085-92. [ Links ]
5. Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002;287:1308-20. [ Links ]
6. Hughes SE. The pathology of hypertrophic cardiomyopathy. Histopathol 2004;44:412-27. [ Links ]
7. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the cardia study. Circulation 1995;92:785-9. [ Links ]
8. Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H. Prevalence of idiopathic hypertrophic cardiomyopathy in China: A populationbased echocardiographic analysis of 8080 adults. Am J Med 2004;116:14-8. [ Links ]
9. Martín M, Rodríguez-Reguero JJ, Calvo D, de la Torre A, Fernández A, García-Castro M, del Valle M y Morís de la Tassa C. Rendimiento del estudio electrocardiográfico en el reconocimiento deportivo de futbolistas federados de una comunidad autónoma. Rev Esp Cardiol 2008;61:426-9. [ Links ]
10. Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A. Sudden death in hypertrophic cardiomyopathy: Identification of high risk patients. J Am Coll Cardiol 2000;36:2212-8. [ Links ]
11. Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J Cardiovasc Electrophysiol 2008;19:104-10. [ Links ]
12. Jaenicke T, Diederich KW, Haas W, Schleich J, Lichter P, Pfordt M. The complete sequence of the human beta-myosin heavy chain gene and a comparative analysis of its product. Genomics 1990;8:194-206. [ Links ]
13. Liew CC, Sole MJ, Yamauchi-Takihara K, Kellam B, Anderson DH, Lin LP. Complete sequence and organization of the human cardiac beta-myosin heavy chain gene. Nucleic Acids Res 1990;18:3647-51. [ Links ]
14. Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE. A molecular basis for familial hypertrophic cardiomyopathy: A beta cardiac myosin heavy chain gene missense mutation. Cell 1990;62:999-1006. [ Links ]
15. Moolman JC, Corfield VA, Posen B, Ngumbela K, Seidman C, Brink PA, Watkins H. Sudden death due to troponin T mutations. J Am Coll Cardiol 1997;29:549-55. [ Links ]
16. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O'Donoghue A. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med 1995;332:1058-64. [ Links ]
17. Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet 1995;11:434-7. [ Links ]
18. Poetter K, Jiang H, Hassanzadeh S, Master SR, Chang A, Dalakas MC. Mutations in either the essential or regulatory light chains of myosin are associated with a rare myopathy in human heart and skeletal muscle. Nat Genet 1996;13:63-9. [ Links ]
19. Seidman JG, Seidman C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001;104:557-67. [ Links ]
20. Charron P, Heron D, Gargiulo M, Richard P, Dubourg O, Desnos M. Genetic testing and genetic counselling in hypertrophic cardiomyopathy: The French experience. J Med Genet 2002;39:741-6. [ Links ]
21. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003;107:2227-32. [ Links ]
22. Keren A, Syrris P, Mc Kenna WJ. Hypertrophic cardiomyopathy: the genetic determinants of clinical disease expression. Nature 2008;5:158-68. [ Links ]
23. Oliveira SM, Ehtisham J, Redwood CS, Ostman-Smith I, Blair EM, Watkins H. Mutation analysis of AMP-activated protein kinase subunits in inherited cardiomyopathies: implications for kinase function and disease pathogenesis. J Mol Cell Cardiol 2003;35:1251-5. [ Links ]
24. Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA 2002;287:1308-20. [ Links ]
25. Charron P, Komajda M. Genes and their polymorphisms in monoand multifactorial cardiomyopathies: Towards pharmacogenomics in heart failure. Pharmacogenomics 2002;3:367-78. [ Links ]
26. Eckart RE, Scoville SL, Campbell CL, Shry EA, Stajduhar KC, Potter RN, Pearse LA, Virmani R. Sudden death in young adults: A 25-year review of autopsies in military recruits. Ann Intern Med 2004;141:829-34. [ Links ]
27. Pelliccia A y Maron BJ. Outer limits of the athlete´s heart: The effect of gender and relevante to the differential diagnosis with primary cardiac diseases. Cardiol Clin 1997;15:381-96. [ Links ]
28. Roeske WR, O´Rourke RAG, Klein A, Leopold G, Karoline JO. Non-invasive evaluation of ventricular hypertrophy in professional athletes. Circulation 1976;53:286-92. [ Links ]
29. Radvan J, Choudhury L, Sheridan DJ, Camici PG. Comparison of coronary vasodilatory reserve in elite rowing athletes versus hypertrophic cardiomyopathy. Am J Cardiol 1997;80:1621-3. [ Links ]
30. Maron BJ, Olivotto I, Spirito P, Casey SA, Bellone P, Gohman TE, Graham KJ, Burton DA, Cecchi F. Epidemiology of hypertrophic cardiomyopathy-related death revisited in a large non-referral based patient population. Circulation 2000;102:858-64. [ Links ]
31. Romeo F, Cianfrocca C, Pelliccia F, Colloridi V, Cistofani R, Reale A. Long-term prognosis in children with hypertrophic cardiomyopathy: An analysis of 37 patients aged less than or equal to 14 years at diagnosis. Clin Cardiol 1990;13:101-7. [ Links ]
32. Koenig H, Goldstone A, Lu CY. Testosterone-mediated sexual dimorphism of the rodent heart. Circ Res 1982;50:782-7. [ Links ]
33. Basavarajaiah S, Boraita A, Whyte G, Wilson M, Carby L, Shah A, Sharma S. Ethnic differences in left ventricular remodeling in highly trained athletes. J Am Coll Cardiol 2008;51:2256-62. [ Links ]
34. Furlanello F, Bettini R, Cozzi F, Del Favero A, Disertori M, Vergara G, Durante GB, Guarnerio M, Inama G, Thiene G. Ventricular arrhythmias and sudden death in athletes. Ann N Y Acad Sci 1984;427:253-79. [ Links ]
35. Pluim BM, Van der Laarse A, Vuelen HW, Bruschke AVG, Van der Wall EE. Left ventricular hypertrophy: Pathology versus physiology. En, Van der Wall EE, Van der Kaarse A, Pluim BM, Bruschke AVG (ed.): Left ventricular hypertrophy: Physiology versus pathology. Kluwer Acad. Publ., Dordrecht, 1999, pp.65-84. [ Links ]
36. Gwathmey JK, Slawsky MT, PerreaultCL, Briggs GM, Morgan JP, Wei JY. Effect of exercise conditioning on excitation-contraction coupling in aged rats. J Appl Physiol 1990;69:1366-71. [ Links ]
37. Rowland T. Sudden unexpected death in young athletes: Reconsidering "hypertrophic cardiomyopathy". Pediatrics 2009;123:1217-22. [ Links ]
38. Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc 2005;80:463-9. [ Links ]
39. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 1996;94:983-91. [ Links ]
40. Pinamonti B, Sinagra G, Salvi A, Di Lenarga A, Morgera T, Silvestri F. Left ventricular involvement in right ventricular dysplasia. Am Heart J 1992;123:711-24. [ Links ]
41. Corrado D, Buja G, Basso C, Thiene G. Clinical diagnosis and management strategies in arrhythmogenic right ventricular cardiomyopathy. J Electrocardiol 2000;33:49-55. [ Links ]
42. Corrado D, Fontaine G, Marcus FI, McKenna WJ, Nava A, Thiene G. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Need for an international registry. Study group on arrhythmogenic right ventricular dysplasia/cardiomyopathy of the working groups on myocardial and pericardial disease and arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation 2000;101:E101-6. [ Links ]
43. Corrado D, Basso C, Schiavon M, Thiene G. Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998;339:364-9. [ Links ]
44. Douglas PS, O'Toole ML, Hiller WDB, Reicheck N. Different effects of prolonged exercise on the right and left ventricles. J Am Coll Cardiol 1990;15:64-9. [ Links ]
45. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomström-Lundquist C, Fontaine G. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J 1994;71:215-8. [ Links ]
46. Tome Esteban MT, García-Pinilla JM, McKenna WJ. Actualización en miocardiopatía arritmogénica del ventrículo derecho: Genética, diagnóstico, manifestaciones clínicas y estratificación de riesgo. Rev Esp Cardiol 2004;57:757-67. [ Links ]
47. Laurent M, Descaves C, Biron Y, Deplace C, Almange C, Daubert JC. Familial occurrence of right ventricular dysplasia. Am Heart J 1987;113:827-9. [ Links ]
48. Severini GM, Krajinovic M, Pinamonti B, Sinagra G, Fioretti P, Brunazzi MC. A new locus for arrhythmogenic right ventricular dysplasia on the long arm of chromosome 14. Genomics 1996;31:193-200. [ Links ]
49. Rampazzo A, Beffagna G, Nava A, Occhi G, Bauce B, Noiato M. Arrhythmogenic right ventricular cardiomyopathy type 1 (arvd1): Confirmation of locus assignment and mutation screening of four candidate genes. Eur J Hum Genet 2003;11:69-76. [ Links ]
50. Rampazzo A, Nava A, Erne P, Eberhard M, Vian E, Slomp P. A new locus for arrhythmogenic right ventricular cardiomyopathy (ARVD2) maps to chromosome 1q42-q43. Hum Mol Genet 1995;4:2151-2154. [ Links ]
51. Awad MM, Calkins H, Judge DP. Mechanisms of disease: Molecular genetics of arrhythmogenic right ventricular dysplasia/ cardiomyopathy. Nat Clin Pract Cardiovasc Med 2008;5:258-67. [ Links ]
52. Rampazzo A, Nava A, Miorin M, Fonderico P, Pope B, Tiso N. Arvd4, a new locus for arrhythmogenic right ventricular cardiomyopathy, maps to chromosome 2 long arm. Genomics 1997;45:259-63. [ Links ]
53. Ahmad F, Li D, Karibe A, Gonzalez O, Tapscott T, Hill R. Localization of a gene responsible for arrhythmogenic right ventricular dysplasia to chromosome 3p23. Circulation 1998;98:2791-5. [ Links ]
54. Li D, Ahmad F, Gardner MJ, Weilbaecher D, Hill R, Karibe A. The locus of a novel gene responsible for arrhythmogenic right-ventricular dysplasia characterized by early onset and high penetrance maps to chromosome 10p12-p14. Am J Hum Genet 2000;66:148-56. [ Links ]
55. Arnemann J, Spurr NK, Magee AI, Buxton RS. The human gene (DSG2) coding for HDGC, a second member of the desmoglein subfamily of the desmosomal cadherins, Is, like DSG1 coding for desmoglein DGI, assigned to chromosome 18. Genomics 1992;13:484-6. [ Links ]
56. Grossmann KS, Grund C, Huelsken J, Behrend M, Erdmann B, Franke WW. Requirement of plakophilin 2 for heart morphogenesis and cardiac junction formation. J Cell Biol 2004;167:149-60. [ Links ]
57. Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet 2004;36:1162-4. [ Links ]
58. Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2002;71:1200-6. [ Links ]
59. Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen- Chowdhry S. Arrhythmogenic right ventricular dysplasia/ cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet 2006;79:978-84. [ Links ]
60. Ferreiro A, Ceuterick-de Groote C, Marks JJ, Goemans N, Schreiber G, Hanefeld F. Desmin-related myopathy with mallory body-like inclusions is caused by mutations of the selenoprotein n gene. Ann Neurol 2004;55:676-86. [ Links ]
61. MacRae CA, Birchmeier W, Thierfelder L. Arrhythmogenic right ventricular cardiomyopathy: Moving toward mechanism. J Clin Invest 2006;116:1825-8. [ Links ]
62. Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, Tjan TD, Soeparwata R, Block M, Borggrefe M, Scheld HH, Breithardt G, Böcker D. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: Single-center experience of long-term follow-up and complications in 60 patients. Circulation 2004;109:1503-8. [ Links ]
63. Buja G, Estes M, Wichter T, Corrado D, Marcus F, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: Risk stratification and therapy. Prog Cardiovasc Dis 2008;50:282-93. [ Links ]
64. Corrado D, Basso C, Sciavon M, Pelliccia A, Thiene G. Pre-participation sports clearance (carta). J Am Coll Cardiol 2009;53:2309-10. [ Links ]
65. Thunenkotter T, Schmied C, Grimm K, Dvorak J, Kindermann W. Precompetition cardiac assessment of football players participating in the 2006 FIFA World Cup Germany. Clin J Sport Med 2009;19:322-5. [ Links ]

