










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkInsuficiencia cardíaca
versão On-line ISSN 1852-3862
Insuf. card. vol.7 no.3 Ciudad Autónoma de Buenos Aires jul./set. 2012
RELATO DE CASO (Versão original)
Cardiomiopatia secundária á distrofia de Steinert
Bruno Afonso Lagoeiro Jorge1, Pedro Gemal Lanzieri1, Fábio Braida do Carmo1, Paula Maíra Alves Haffner2, Antônio José Lagoeiro Jorge3, Wolney de Andrade Martins4
1 Acadêmico de graduação em Medicina. Universidade Federal Fluminense (UFF). Niterói (RJ). Brasil.
2 Médica Residente em Cardiologia. Universidade Federal Fluminense (UFF). Niterói (RJ). Brasil.
3 Mestre em Ciências Cardiovasculares. Universidade Federal Fluminense (UFF). Niterói (RJ). Brasil.
4 Doutor em Cardiologia. Universidade de São Paulo. São Paulo. Brasil.
Professor da Faculdade de Medicina. Universidade Federal Fluminense (UFF). Niterói (RJ). Brasil.
Instituição: Universidade Federal Fluminense (UFF). Departamento de Medicina Clínica. Niterói (RJ). Brasil.
Correspondência: Dr. Bruno Afonso Lagoeiro Jorge
Avenida Marques do Paraná 303. Sexto andar. CEP: 24030-215. Centro de Cardiologia. Niterói. (RJ). Brasil.
Telefone: 552126299207
E-mail: brunolagoeiro@hotmail.com
O autor e os coautores estão de acordo com todo o conteúdo expresso no manuscrito e assumem a responsabilidade pela veracidade do relato.
Este trabalho é parte do Diretório de pesquisa "Insuficiência cardíaca da molécula à população" do curso de Pós-Graduação em Ciências Cardiovasculares da Universidade Federal Fluminense (UFF).
Recebido: 02/02/2012
Aceitado: 20/06/2012
Resumo
A distrofia miotônica tipo 1 (DM1 ou doença de Steinert) é uma doença genética com repercussões multissistêmicas, na qual é comum o paciente buscar diversos especialistas antes da suspeição clínica. O envolvimento cardíaco é uma das características principais da evolução da DM1 e explica, em parte, a menor expectativa de vida dos pacientes. Relata-se caso de paciente masculino, 28 anos, com admissão por insuficiência cardíaca descompensada após oito anos de diagnóstico quando apresentou episódio de síncope secundária a bloqueio atrioventricular total. Sob marca-passo definitivo desde então. Discutem-se os aspectos etiológicos, fisiopatológicos e clínicos da DM1 com ênfase nas manifestações cardiovasculares.
Palavras-chave: Doença de Steinert; Distrofia miotônica; Insuficiência cardíaca; Bloqueio atrioventricular; Marca-passo artificial.
Introdução
A distrofia miotônica (DM) progressiva é uma síndrome genética, autossômica dominante, de herança materna e é a doença neuromuscular mais freqüente em adultos. Tem prevalência de 1:8000 nascimentos1 e se apresenta como tipo 1 (DM1 ou Distrofia de Steinert) e tipo 2 (DM2). A DM1 é o tipo mais comum e se manifesta, em sua forma clássica, entre os 12 e 30 anos de idade. Os pacientes apresentam anormalidades cardíacas como distúrbios da condução atrioventricular; arritmias atriais e ventriculares; e insuficiência cardíaca (IC). Outros acometimentos são hipogonadismo e infertilidade, catarata, distúrbios do sono, resistência insulínica e hipotireoidismo. Devido ao quadro multissistêmico, é comum o paciente buscar diversos especialistas antes da suspeição clínica da doença.
Relato do caso
História e exame físico
SRCO, 28 anos, natural de Niterói, Rio de Janeiro, Brasil, solteiro e professor. Diagnosticado com distrofia de Steinert em 2003, após investigação de episódio sincopal, que culminou no diagnóstico de bloqueio atrioventricular (BAV) total e conseqüente implante de marca-passo (MP), dupla câmara. Evoluiu assintomático nos cinco anos seguintes. Em 2008, iniciou quadro de dispneia aos grandes esforços, ocasião em que foi diagnosticada IC. Foi iniciado tratamento ambulatorial com carvedilol, espironolactona, furosemida e digoxina. Em 2011, apresentou piora clínica, com agravamento dos sintomas neuromusculares e sinais de congestão sistêmica. Há três meses da internação, apresentou dispneia progressiva, que evoluiu dos grandes aos mínimos esforços (classe funcional II para III da New York Heart Association -NYHA-), ortopnea, dispneia paroxística noturna (DPN), edema em membros inferiores e aumento do volume abdominal. Mãe teve diagnóstico de DM1 e cardiopatia, com óbito aos 33 anos de idade. Sem outros casos doenças cromossômicas e morte súbita na família.
Ao exame físico: lúcido e orientado; fácies típica da doença de Steinert, com atrofia dos músculos temporal e masseter. Frequência cardíaca de 60bpm; pressão arterial de 90/60 mm Hg. Emagrecido (peso= 50,7kg e índice de massa corporal= 17,1 Kg/m2). Taquipneico (22 irpm), sem esforço respiratório. Ausência de turgência jugular à 45 graus. Ictus cordis visível no 5º espaço intercostal, na linha hemiclavicular esquerda, globoso, duas polpas digitais. Ritmo cardíaco regular em quatro tempos por 3ª e 4ª bulhas, sem sopros. Ginecomastia bilateral, simétrica, indolor. Circulação colateral venosa em face anterior do tórax. Musculatura intercostal consumida, com arcos costais visíveis. Exame pulmonar sem alterações. Abdome com circunferência de 76cm e presença de ascite moderada. Membros inferiores com edema (3+/4+). Pupilas anisocóricas (esquerda>direita) e fotorreagentes. Ausência de reflexo miotônico. Estática e coordenação preservadas. Força muscular grau 4,0 proximal nos membros superiores, inferiores e pescoço, e 4- nos pés. Reflexos profundos hipoativos e simétricos. Sensibilidade preservada. (Figuras 1A e 1B).

Figura 1. Fotografias do frente (A) e perfil (B) de paciente com Doença de Steinert. Evidenciam atrofia muscular, ginecomastia
secundária à espironolactona, ascite e gerador de marca-passo.
Exames Complementares
Telerradiografia do tórax em posteroanterior e perfil evidenciaram cardiomegalia (índice cardiotorácico= 0,53); MP dupla câmara; e pulmões sem alterações (Figuras 2A e B). Eletrocardiograma mostrou comando de marca-passo (Figura 3). O ecocardiograma evidenciou aumento biatrial; disfunção biventricular com hipocinesia difusa grave do ventrículo esquerdo; regurgitação mitral leve e tricúspide moderada; pressão sistólica de artéria pulmonar estimada em 48 mm Hg; veia cava inferior congesta; e dissincronismo atrioventricular. Clearance de creatinina estimado= 96 mL/min, Na+=130 mEq/L, bilirrubina total= 2,89 mg/dL, com fração direta= 1,69 mg/dL.

Figura 2. Telerradiografia do tórax em posteroanterior (A) e perfil (B). Índice cardiotorácico de 0,53 e marca-passo.
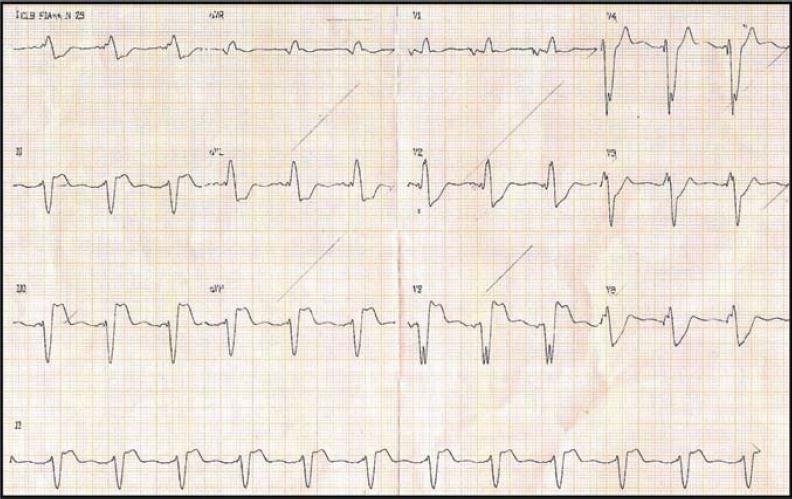
Figura 3. Eletrocardiograma basal da internação mostra comando de marca-passo.
Evolução
Foi admitido na enfermaria de cardiologia e iniciou-se furosemida venosa, carvedilol, espironolactona e digoxina, que posteriormente foi suspensa devido à elevação da digoxinemia (3,5 ng/mL). Paciente permaneceu internado por oito dias, com melhora clínica e laboratorial. Retornou à classe funcional II da NYHA, com remissão da DPN. Houve regressão do edema de membros inferiores para (1/4+); redução discreta da ascite e do volume abdominal (74 cm); e diminuição do peso corporal com perda de 1,7 Kg. Recebeu alta com melhora dos sintomas e regressão dos sinais.
Discussão
A apresentação clínica da DM1 varia de acordo com o número de repetições da cadeia citosina-tirosinaguanina (CTG) no gene da proteína-quinase. O número de repetições determina a ocorrência das formas clínicas branda (50-150 repetições), clássica (100-1000) ou grave (500-2700)1. Os doentes com perfil clássico de DM1 têm início dos sintomas entre os 12 e 30 anos de idade, apresentando-se com fraqueza e atrofia muscular; miotonia; catarata; alopecia; e distúrbios da condução cardíaca1. Embora não tenha sido realizada a pesquisa genética, o paciente desse relato apresentou história e manifestações clínicas sugestivas do fenótipo clássico, ou seja, alteração da marcha; preservação da musculatura das mãos; fácies típica, incluindo ptose palpebral e atrofia da musculatura do masseter; e disfagia para sólidos. Na DM1 predomina a fraqueza muscular distal progressiva, com dificuldade de realizar movimentos finos com mãos e pés. A fácies característica depende da fraqueza da musculatura facial. Os músculos flexores da nuca são comumente envolvidos. Ocorre lesão dos neurônios adrenérgicos da formação reticular ascendente, que pode ser acompanhada de neuropatias2. Acometimento do trato gastrointestinal desencadeia redução da motilidade da hipofaringe e do esôfago proximal, com consequente disfagia e pneumonias por aspiração1-5. Outras características clínicas descritas, porém ausentes nesse paciente, são: catarata, alopecia, resistência insulínica, síndrome do intestino irritável, colecistite e miotonia1.
O paciente relatado teve apresentação clínica da DM1 aos 20 anos de idade e referiu óbito da mãe pela mesma doença aos 33 anos. Estudos demonstram que a expectativa de vida desses pacientes está abaixo da média população geral. As causas imediatas de mortalidade, em ordem decrescente, são: (1ª) pneumonia; (2ª) falência neuromuscular; (3ª) doença cardiovascular ou morte súbita atribuída aos distúrbios de condução; e (4ª) câncer6-8. A média da idade de mortalidade referida na literaturaé 54 anos, com relatos de sobrevida até os 80 anos, com maior longevidade atribuída à forma branda da doença. A expectativa de vida na DM1 difere das demais doenças neuromusculares como Duchenne que possuem expectativa vida significativamente menor. Infecções de repetição e complicações tromboembólicas aumentam o risco de mortalidade desses pacientes9-11.
O paciente deixou de exercer suas atividades laborais há dois anos da presente internação. As manifestações clínicas da DM1 podem afetar o estado comportamental, emocional e cognitivo, o que dificulta a interação social e o grau de aprendizado. Isso prejudica as atividades diárias e reduz a qualidade de vida. Em pacientes com DM1, o comprometimento do estado mental e a presença de distúrbios comportamentais são fatores que influenciam o manejo e a terapêutica e, consequentemente no prognóstico12, o que não esteve presente neste caso.
O envolvimento cardíaco é uma das características principais da evolução da DM1. A histopatologia cardíaca pode demonstrar fibrose do sistema de condução e do nodo atrioventricular, hipertrofia dos cardiomiócitos e infiltração gordurosa. Há bandas I proeminentes e degeneração miofibrilar à microscopia eletrônica. Fibrose miocárdica e degeneração do sistema de condução podem levar a bloqueio atrioventricular, assistolia, taquiarritmia ventricular ou morte súbita11. São freqüentes os distúrbios de condução, principalmente no sistema de His-Purkinje, com destaque para o aumento do intervalo PR e o alargamento do QRS. Apesar da elevada incidência, essas alterações costumam ser subclínicas13. Arritmias atriais ou ventriculares podem estar presentes, geralmente benignas. Há marcada dissociação entre a prevalência de alterações eletrocardiográficas com a baixa frequência de sintomas cardiovasculares14. Não há correlação estabelecida entre o grau de sintomas cardiovasculares e musculoesqueléticos. Acredita-se que, na forma clássica, quanto mais precoce ocorrerem as manifestações neuromusculares – ainda que apenas a miotonia na segunda década de vida – maior será o comprometimento do sistema His-Purkinje. Por outro lado, quando o início dos sintomas neuromusculares for mais tardio, entre a terceira e quarta décadas, os distúrbios da condução cardíaca provavelmente se correlacionarão com o tempo de evolução e o grau de variação fenotípica da doença. A presença de distúrbio da condução cardíaca no caso relatado é considerada fator de pior prognóstico13.
Ainda não foram estabelecidos os critérios de elegibilidade para implante de MP nesses pacientes, mas foi demonstrado seu uso precoce pode evitar a morte súbita. A cardiomiopatia dilatada pode ser precoce, porém assintomática, e a intolerância aos esforços é um parâmetro tardio de apresentação da IC, já que a restrição, pela disfunção musculoesquelética, impede a prática de exercícios vigorosos por esses pacientes. A proporção de portadores de DM1 e IC com fração de ejeção preservada ainda não foi descrita na literatura. A etiopatogenia da disfunção miocárdica ocorre por falência muscular pura ou é agravada pelo dissincronismo presente nestes pacientes. É provável que a presença de MP sem função de ressincronização possa agravar a dissincronia ventricular e, consequentemente, piorar a disfunção cardíaca. A doença de Steinert deve ser levada em consideração como diagnóstico diferencial em pacientes jovens com quadro de síncope associada a BAV. Além disso, pacientes diagnosticados devem ser acompanhados com ECG seriados15.
A pesquisa genética no diagnóstico da DM1 avançou a partir de 1992, com a disponibilização do teste de biologia molecular. Diversos estudos foram publicados apresentando a expansão instável de repetição da cadeia CTG no gene da proteína-quinase16-20, hoje considerado o padrão ouro para o diagnóstico de DM1. É apropriado iniciar a confirmação diagnóstica com a análise genética, em vez da eletroneuromiografia (ENM). Em geral, as informações da ENM são limitadas nesses pacientes, entretanto pode ser útil para casos atípicos em que não há clínica aparente de miotonia ou quando os testes moleculares para DM são normais. No caso relato, o diagnóstico de DM1 foi baseado na impressão clinica, prática com respaldo na literatura. Usualmente, este diagnóstico pode ser estabelecido por fraqueza muscular e miotonia, somados à história familiar positiva. O paciente relatado apresentou, inicialmente, um distúrbio de condução cardíaca, embora já tivesse o critério da história familiar positiva e o quadro neuromuscular. O tratamento da DM1 baseia-se em medidas sintomáticas e apoio multidisciplinar. São empregadas próteses ortopédicas, cirurgias oculares, fisioterapia motora e fármacos para controle da mialgia tais como os antiinflamatórios, gabapentina, antidepressivo, metilxantinas e glicocorticóides em baixas doses2.Como os sintomas neuromusculares foram mais brandos no paciente relatado, não se aplicou tal terapêutica.
Conclusão
A DM1 é doença grave com comprometimento cardiovascular freqüente. Os pacientes devem ser monitorados quanto às lesões cardiovasculares, em especial a cardiomiopatia e os distúrbios de condução, freqüentes nesses pacientes e que podem ser subestimados frente à magnitude dos sintomas neuromusculares. Ainda que assintomáticos do ponto de vista cardiovascular, são recomendados exames de rastreio para diagnóstico precoce dessas condições, embora o prognóstico seja reservado na maioria dos casos.
Recursos financeiros
A linha de pesquisa em insuficiência cardíaca e cardiomiopatias do Curso de Pós-Graduação em Ciências Cardiovasculares da UFF contam com fomento da Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ).
Conflito de interesses
Não há conflitos de interesses pertinentes.
1.Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry 2010;81:358-67. [ Links ]
2. Krishnan AV, Kiernan MC. Axonal function and activity-dependent excitability changes in myotonic dystrophy. Muscle Nerve 2006;33:627-36. [ Links ]
3. Ronnblom A, Forsberg H, Danielsson A. Gastrointestinal symptoms in myotonic dystrophy. Scand J Gastroenterol 1996;31:654-7. [ Links ]
4. Ronnblom A, Andersson S, Hellstrom PM, Danielsson A. Gastricemptying in myotonic dystrophy. Eur J Clin Invest 2002;32:570-4. [ Links ]
5. Heatwole CR, Miller J, Martens B, Moxley RT. Laboratory abnormalities in ambulatory patients with myotonic dystrophy type 1. Arch Neurol 2006;63:1149-53. [ Links ]
6. Reardon W, Newcombe R, Fenton I, Sibert J, Harper PS. The natural history of congenital myotonic dystrophy: mortality and long term clinical aspects. Arch Dis Child 1993;68:177-81. [ Links ]
7. Schoser BG, Ricker K, Schneider-Gold C, Hengstemberg C, Durre J, Bultmann B et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology 2004;63:2402-4. [ Links ]
8. Nazarian S, Wagner KR, Caffo BS, Tomaselli GF. Clinical predictors of conduction disease progression in type I myotonic muscular dystrophy. Pacing Clin Electrophysiol 2011;34:171-6. [ Links ]
9. De Die-Smulders CE, Höweler CJ, Thijs C, Mirandolle JF, Anten HB, Smeets HJM et al. Age and causes of death in adult-onset myotonic dystrophy. Brain 1998;121(8):1557-63. [ Links ]
10. Mathieu J, Allard P, Potvin L, Prévost C, Bégin P. A 10-year study of mortality in a cohort of patients with myotonic dystrophy. Neurology 1999;52:1658-62. [ Links ]
11. Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med 2008;358:2688-97. [ Links ]
12. Roig M, Balliu PR, Navarro C, Brugera R, Losada M. Presentation, clinical course, and outcome of the congenital form of myotonic dystrophy. Pediatr Neurol 1994;11:208-13. [ Links ]
13. Nishioka SAD, Filho MM, Marie S, Zatz M, Costa R. Distrofia miotônica e cardiopatia. Comportamento dos eventos arrítmicos e dos distúrbios da condução. Arq Bras Cardiol 2005;84(4):330-6. [ Links ]
14. Rodríguez IJP, Rivas EG, Cabello A. Repercusión cardíaca de las enfermedades neuromusculares. Rev Esp Cardiol 1997;50:882-901. [ Links ]
15. Melacini P, Buja G, Fasoli G, Angelini C, Armani M, Scognamiglio R et al. The Natural History of Cardiac Involvement in Myotonic Dystrophy: An Eight-Year Follow-Up in 17 Patients. Clin Cardiol 1998:11:231-8. [ Links ]
16. Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992;69:799-808. [ Links ]
17. Buxton J, Shelbourne P, Davies J, Jones C, Tongeren TV, Charalampos A et al. Detection of an unstable fragment of DNA specific to individuals with myotonic dystrophy. Nature 1992;355:547-48. [ Links ]
18. Fu YH, Pizzuti A, Fenwick RG Jr, King J, Rajnarayan S, Dunne PW. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256-58. [ Links ]
19. Harley HG, Brook JD, Rundle SA, Crow S, Reardon W, Buckler AJ et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature 1992;355:545-6. [ Links ]
20. Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Janses G et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 1992;255:1253-55. [ Links ]
21. Kurihara T. New classification and treatment for myotonic disorders. Intern Med 2005;44:102. [ Links ]

