










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. vol.9 no.2 Ciudad Autónoma de Buenos Aires jun. 2014
ARTÍCULO DE REVISIÓN
Síndrome antifosfolipídico y afectación cardiovascular
Diego D. Zanazzi1
1 Médico cardiólogo. Servicio de Cardiología. Hospital "Julio C. Perrando". Resistencia. Chaco. República Argentina.
Correspondencia: Dr. Diego D. Zanazzi.
Servicio de Cardiología. Hospital "Julio C. Perrando".
9 de Julio 1100. CP: 3500. Resistencia. Chaco. República Argentina.
E-mail: diego_z_1983@hotmail.com
Recibido: 16/09/2013
Aceptado: 10/02/2014
Resumen
El síndrome antifosfolípido se define como la presencia de trombosis arteriales o venosas recurrentes, pérdidas repetidas de embarazo y trombocitopenia en presencia de anticuerpos a cardiolipina o anticoagulante lúpico. Se realizo esta revisión con el fin de determinar si este síndrome debe ser considerado actualmente un factor de riesgo cardiovascular. Se presenta en mujeres jóvenes por lo cual al ser diagnosticado nos obliga a realizar un control exhaustivo y debe tener un abordaje multidisciplinario. Dentro de las manifestaciones cardiovasculares el paciente puede presentar infarto agudo de miocardio, endocarditis pseudo-infecciosa, afectación valvular e hipertensión pulmonar.
Palabras clave: Síndrome antifosfolipídico ; Anticuerpos antifosfolípidos ; Afectación cardiovascular ; Hipertensión pulmonar
Summary
Antiphospholipid syndrome and cardiovascular affectation
Antiphospholipid syndrome is defined as the presence of recurrent arterial or venous thrombosis, repeated pregnancy loss and thrombocytopenia in the presence of antibodies to cardiolipin or lupus anticoagulant. This review was performed in order to determine whether this syndrome should now be considered a cardiovascular risk factor. It occurs in young women, therefore, to be diagnosed forces us to make a thorough control and should have a multidisciplinary approach. Among the cardiovascular manifestations the patient may develop acute myocardial infarction, pseudo-infectious endocarditis, valve disease and pulmonary hypertension.
Keywords: Antiphospholipidic syndrome ; Antiphospholipid antibodies ; Cardiovascular affectation ; Pulmonary hypetension
Resumo
Síndrome do anticorpo antifosfolipídeo e comprometimento cardiovascular
A síndrome do anticorpo antifosfolipídeo é definida como a presença de trombose arterial ou venosa recorrente, repetidos abortos e trombocitopenia na presença de anticorpos anti-cardiolipina ou lúpus anticoagulante. Esta avaliação foi realizada, a fim de determinar se esta síndrome deve agora ser considerada um fator de risco cardiovascular. Ela ocorre em mulheres jovens, por isso, de ser diagnosticada nos obriga a fazer um estudo aprofundado e deve ter uma abordagem multidisciplinar. Entre as manifestações cardiovasculares, o paciente pode desenvolver infarto agudo do miocárdio, endocardite pseudo-infecciosa, valvopatia e hipertensão pulmonar.
Palavras-chave: Síndrome do anticorpo antifosfolipídeo ; Anticorpo antifosfolipídeo ; Comprometimento cardiovascular ; Hipertensão pulmonar
Introducción
El síndrome antifosfolipídico (SAF), también, es conocido como "síndrome de la sangre pegajosa" o "síndrome de Hughes" en honor al Dr. Graham R. V. Hughes, reumatólogo del Hospital de St. Thomas de Londres (Reino Unido), quien lo describió por primera vez en 19831. Es una enfermedad rara, sistémica y de características autoinmunes, que provoca un estado trombofílico, debido a la presencia en sangre de anticuerpos dirigidos contra el complejo protrombina-fosfolípidos o contra el factor de coagulación-fosfolípidos que afectan la conversión de la primera en trombina y la activación de los segundos2,3.
El SAF es un síndrome que se presenta sobre todo en mujeres entre 20 y 40 años de edad4, caracterizándose por abortos espontáneos (con mayor frecuencia en el primer trimestre del embarazo), trombosis venosa profunda (TVP) y trombocitopenia. Presenta serológicamente anticuerpos antifosfolípidos y/o anticoagulantes lúpicos. El 55% de los pacientes con el síndrome manifiesta TVP, en especial en los miembros inferiores, la mitad de los cuales tiene además embolia pulmonar5. La afectación cardíaca en este síndrome es poco conocida, consecuentemente, la presente revisión está destinada a dar a conocer sus implicancias cardiovasculares.
Historia
En 1906, Wassermann fue el primero en describir la presencia de anticuerpos antifosfolípidos en pacientes con serología positiva para sífilis6,7, los cuales se llamaron anticoagulantes lúpicos (AL), aunque sólo muchos años después se identificó a la cardiolipina (un fosfolípido mitocondrial) como el antígeno más importante en el SAF. Pronto, se observó que a muchos pacientes con lupus eritematoso sistémico (LES) les daba positivo la prueba de VDRL (por su siglas en inglés, Venereal Disease Research Laboratory, que usa como sustrato un antígeno compuesto de cardiolipina-fosfatidilcolina y colesterol para detectar sífilis), sin evidencia clínica ni serológica de sífilis. En 1983, se desarrolló un inmunoensayo mucho más sensible para la detección de anticuerpos anticardiolopinas y pronto se encontró una fuerte correlación entre los anticuerpos asociados al LES8,9 y la trombosis10.
Eon Nigel Harris y Graham R. V. Hughes fueron los primeros en formalizar el concepto de síndrome de anticuerpos antifosfolípidos, en 1980; siendo Hughes quien lo describe por primera vez1,10,11. El SAF ya era considerado una entidad clínica definida que podía existir por sí sola (SAF primario) o asociada a otras patologías autoinmunes (SAF secundario).
Hacia 1990, dos grupos independientes descubrieron que algunos anticuerpos anticardiolopinas necesitaban la presencia de una proteína plasmática capaz de unirse a los fosfolípidos plasmáticos: denominada beta 2 glucoproteína I (B2-GPI). Ésta se fija a los fosfolípidos aniónicos e inhibe la vía intrínseca de las plaquetas11. Los anticuerpos anticardiolipinas al necesitar de este cofactor interfieren con las acciones de la B2-GPI, favoreciendo los fenómenos trombóticos, por ello la trombocitopenia es frecuente en el SAF, pero a su vez no tan severa como para producir hemorragias3,4.
En 1999, se realizan los criterios diagnósticos oficiales del SAF (criterios de Sapporo) que, posteriormente, se modifican en 20063.
Los anticuerpos clínicamente relevantes se clasifican en 3 subgrupos de acuerdo con el método analítico seguido para su detección:
- Anticuerpo anticoagulante lúpico.
- Anticuerpos anticardiolipinas.
- Anticuerpo anti B2-GPI.
Inmunología
Los fosfolípidos son los principales componentes de las membranas celulares y el mecanismo de trombosis obedece a los anticuerpos antifosfolípidos (AAF) que inhiben la reacción en la cascada de la coagulación, catalizado por los fosfolípidos cargados negativamente.
Anticuerpos antifosfolípidos
Los AAF son, fundamentalmente, B2-GPI (Figura 1) y protrombina. Provocan la aparición de trombosis, además de actuar directamente sobre el endotelio vascular12, componiéndose de dos anticuerpos: los anticuerpos anticardiolipina (AAC) y los anticuerpos anticoagulante lúpicos (AAL).

Figura 1. Los anticuerpos anti beta 2 glucoproteína I (B2-GPI) en complejo con la glucoproteína B2-GPI interactúan con ciertos receptores de la superficie de las membranas que poseen fosfolípidos. La B2-GPI es una glucoproteína altamente glucosilada con 5 dominios, que interacciona con los fosfolípidos de membrana a través de su dominio V, rico en lisina. A partir de linfocitos B de pacientes con síndrome antifosfolipídico (SAF) se han obtenido anticuerpos anti B2-GPI.
Los principales antígenos diana en pacientes con SAF incluyen: B2-GPI13, protrombina y anexina V (Figura 2 y 3).

Figura 2. Representación esquemática de cómo los anticuerpos anti beta 2 glucoproteína I (B2-GPI) en complejo con la glucoproteína B2-GPI pueden interactuar con ciertos receptores de la superficie en las plaquetas y células endoteliales para inducir la activación celular.

Figura 3. Las posibles vías intracelulares plaquetaria activadas por anticuerpos antifosfolípidos (aPL). TRAP: péptido agonista del receptor de la trombina (thrombin receptor agonist peptide). PAR: receptor de la proteína activada (proteinactivated receptor). Gp: glucoproteína. Rc: receptor. PLCß: fosfolipasa Cβ. PLCγ: fosfolipasa Cγ. DG: diacilglicerol. IP3: inositol trifosfato. PKC: proteína quinasa C. cPLA2: calcio dependiente de fosfolipasa citosólica A2. AA: ácido acetilsalicílico. TXB2: tromboxano B2. TXRc: receptor del tromboxano. TR: trombina.
Anticuerpos anticoagulantes lúpicos
Los AAL interfieren con las pruebas de coagulación dependientes de fosfolípidos in vitro14, tienen propiedad de alargar los tiempos de coagulación dependiente de fosfolípidos. Prolongan el tiempo de cefalina activada; así, los pacientes con AAL tienen tendencia a presentar fenómenos trombóticos y no hemorrágicos.
Anticuerpos anticardiolipinas
Los AAC se determinan por medio del método de ELISA. Para que la unión de los AAC a su antígeno se produzca, es necesaria la presencia de un cofactor plasmático: la B2-GPI, estando asociados a procesos trombóticos14.
Patogenia
Hay propuestas varias teorías acerca de la patogenia de este síndrome, para explicar los mecanismos celulares y moleculares a través de los cuales los AAF provocan la trombosis15,16.
La primera implica a las células endoteliales que son activadas por la unión de AAF, incrementando la producción de moléculas de adhesión y la secreción de citoquinas que aumentan el metabolismo de las prostaglandinas.
La segunda se enfoca en el daño del endotelio vascular mediado por agentes oxidantes: la lipoproteína de baja densidad (LDL: Low Density Lipoprotein) oxidada es fagocitada por los macrófagos, con su subsecuente activación y daño a la célula endotelial, formándose auto-anticuerpos contra LDL y anticardiolipina, y de éstos algunos reaccionan en forma cruzada con LDL; además la anticardiolipina se fija a cardiolipina oxidada, pero no reducida, sugiriendo que los anticuerpos anticardiolipina reconocen a los fosfolípidos oxidados.
Y la tercera propone que los AAF interfieren o modulan la función de las proteínas de unión a fosfolípidos involucradas con la regulación de la coagulación. La B2-GPI es un anticoagulante natural, uniéndose a fosfolípidos aniónicos inhibe la vía intrínseca de la coagulación y la agregación plaquetaria. Los anticuerpos antifosfolipídicos necesitan a la B2-GPI por lo cual interferirían en su acción favoreciendo los fenómenos trombóticos17.
También, se han propuesto otros mecanismos según los cuales los AAF interferirían sobre la función de la protrombina, proteína C, anexina V y factor V. Finalmente, la trombosis en el SAF podría ser similar a la trombocitopenia inducida por la heparina, según comparan algunos investigadores; además se ha sugerido que los AAF presentes en la superficie externa de la membrana plaquetaria causan daños en las plaquetas, provocando un aumento de la captación y destrucción por el sistema retículo endotelial con acortamiento de su supervivencia.
La regulación de la hemostasia es compleja y ocurre en distintos niveles; destacándose el rol de células endoteliales y las plaquetas; así como de la cascada de la coagulación y fibrinolisis. Existe evidencia actual que señala la habilidad de AAF de interferir en todos los niveles18-20 (Figura 4).

Figura 4. Mecanismos involucrados en la trombosis. B2-GPI: beta 2 glucoproteína I.
Fisiopatología
En cuanto a la fisiopatología del SAF, se ha sugerido que el mecanismo de la trombosis obedece a los AAF que inhiben la reacción en la cascada de la coagulación catalizada por los fosfolípidos cargados negativamente; además, se presenta una interacción entre éstos y activadores antigénicos sobre las plaquetas, células endoteliales y componentes de la cascada de la coagulación que afectan la activación del factor X (Figura 5), la conversión protrombína-trombina, activación de la proteína C y la activación del factor Va, induciendo un estado protrombótico que podría afectar igualmente la síntesis de tromboxano por las plaquetas, inhibiendo la síntesis de prostaciclina y activando células endoteliales de adhesión, la producción de factor tisular por células endoteliales como la endotelina-1, activando la secreción de citoquinas proinflamatorias y la modulación del metabolismo del ácido araquidónico que afecta la proteína S. La hipótesis del segundo hit (golpe) postula que un defecto subyacente endotelial en la presencia de AAF dispara las complicaciones trombóticas21,22.

Figura 5. Acción de anticuerpos antifosfolípidos (aFL) autoinmunes sobre los mecanismos antitrombóticos que controlan la actividad del factor Xa.
En presencia de los anticuerpos anticardiolipina, aumenta la captación de la LDL oxidada, que es vital en todo proceso aterotrombótico23. Los anticuerpos contra la B2-GPI son más frecuentes asociados con las manifestaciones trombóticas (Figura 6) y se componen de dos subtipos de anticuerpos, uno que se une a superficies aniónicas y otros a superficies no cargadas; últimamente, este anticuerpo contra la B2-GPI se ha asociado con las manifestaciones neurológicas del síndrome24,25.
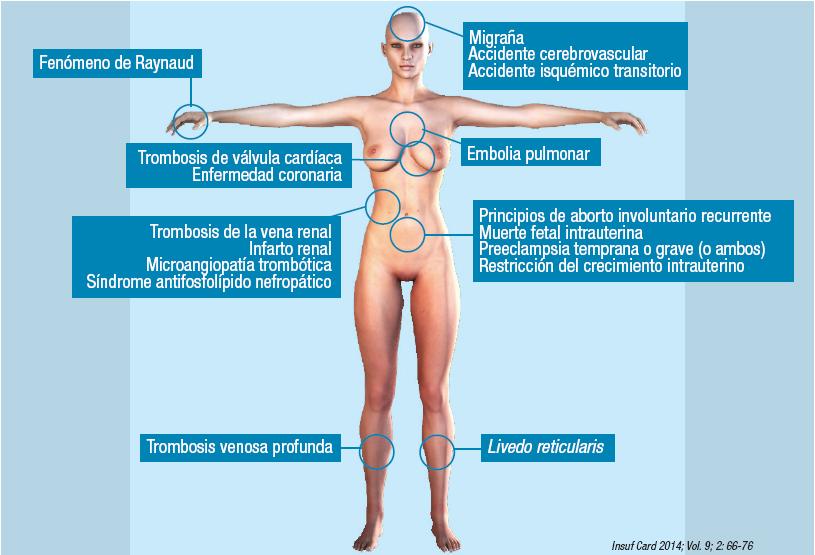
Figura 6. Manifestaciones clínicas del síndrome de anticuerpos antifosfolípidos.
Se ha descrito la interacción de los anticuerpos con la anexina V (proteína I anticoagulante placentaria), por lo que la disminución favorece los eventos trombóticos26,27, esta proteína actúa como tromborreguladora y al ser desplazada por los anticuerpos se desencadena el proceso trombótico, describiéndose igualmente defectos en la placentación28-30, que traducen una insuficiencia placentaria, conduciendo a la aparición de abortos espontáneos de más de 10 semanas, partos prematuros, muertes fetales inexplicadas y crecimiento intrauterino retardado.
En la patogenia del SAF, se viene presentando evidencia de un papel importante del factor de necrosis tumoral alfa como una sustancia protrombótica y proinflamatoria que al detectarse un polimorfismo de su gen puede desencadenar las manifestaciones32.
En enfermedades infecciosas se ha descrito la inducción de AAF con complicaciones como embolia pulmonar33.
Diagnóstico
Para el diagnóstico positivo se deben cumplir los criterios de Saporo 20063.
El diagnóstico positivo del SAF requiere al menos de uno de los criterios clínicos con al menos de uno de los criterios fisiopatológicos de laboratorio.
Criterios clínicos (Tabla 1)
Tabla 1. Criterios clínicos del síndrome de anticuerpos antifosfolípidos

1.- Trombosis vascular: arterial, venosa profunda, capilar, confirmada por imagen o histopatología.
2.- Complicaciones obstétricas: 3 abortos espontáneos antes de las 10 semanas de amenorrea consecutiva o inexplicada. Una muerte fetal in útero inexplicada con feto con morfología normal o un parto prematuro con feto de morfología normal por preeclampsia severa, eclampsia o insuficiencia placentaria.
Criterios fisiopatológicos de laboratorio
1.- Anticuerpos anticoagulante lúpico circulante.
2.- Anticuerpos anticardiolipinas: Anticuerpos anticardiolipinas IgG o IgM presentes con niveles medios o elevados en sangre determinados por ELISA.
3.- Anticuerpos anti B2-GPI IgG o IgM determinado por ELISA.
Manifestaciones cardiológicas
El SAF produce muchas complicaciones sobre el sistema cardiovascular y en particular sobre el corazón, partiendo del hecho de que existen múltiples investigaciones que ponen en evidencias al corazón como órgano blanco en este síndrome, y a la vez como determinante para cardio-embolismos sistémicos.
El SAF afecta al corazón de diferentes formas, pudiendo producir (Tabla 2): a nivel valvular (endocarditis pseudo-infecciosa, valvulopatías con engrosamiento valvular, etc.), como factor independiente de cardiopatía isquémica (con y sin infarto agudo de miocardio [IAM], trombosis de la microcirculación, oclusión de los puentes revascularizados, trombos intracardíacos y cardiomiopatía)5,34,35, e hipertensión pulmonar.
Tabla 2. Manifestaciones cardiovasculares del síndrome de anticuerpos antifosfolípidos (SAF)

Compromiso valvular
A través de estudios ecocardiográficos de muchos reportes de investigadores se evidencia que en el SAF existen 2 tipos de afecciones valvulares: por un lado produce masas (vegetaciones) valvulares y, por otro, engrosamiento valvular.
Estas alteraciones pueden estar combinadas y a su vez asociarse con disfunción valvular, la principal anormalidad funcional es la regurgitación, mientras que las estenosis son muy raras. Las válvulas comúnmente afectadas son la mitral y la aórtica13, con mayor proporción de la primera, ya que ésta presenta una superficie de mayor vulnerabilidad producto del stress-jet y turbulencia a la que está sometida. Por ecocardiografía tienen una localización predominantemente sobre la porción media y proximal de la cúspide valvular. La presencia de afecciones en las válvulas pulmonares y tricúspides es muy poco frecuente36-41.
Los resultados de algunos de los estudios clínicos sugieren que la tercera parte de los pacientes con SAF primario tienen lesiones valvulares en relación a la población general.
Se ha demostrado una prevalencia altamente significativa de compromiso valvular en los pacientes con SAF secundario a LES, en relación al SAF primario y a la población control.
Las lesiones valvulares verrugosas también llamadas de Lidman-Sack son vegetaciones fibrofibrinosas estériles que se pueden desarrollar sobre la superficie del endocardio del corazón con predilección sobre las válvulas izquierdas y en especial sobre la superficie ventricular de la válvula mitral, estas vegetaciones son sésiles y pequeñas de 3 a 4 mm42-50.
Desde el punto de vista ecocardiográfico las vegetaciones aparecen como masas valvulares de forma y tamaño variable unidas firmemente a la superficie sin movilidad independiente y datos ecocardiográficos recientes demuestran una localización predominante sobre la porción media y proximal de la cúspide de la valva de la válvula 37-40. Las lesiones valvulares pueden tener depósito de fibrina, neovascularización y una extensión variable de inflamación con infiltración de células mononucleres42-53.
En cuantos a las manifestaciones clínicas de lesiones valvulares asociadas al SAF en la mayoría de los pacientes, el compromiso valvular fue hemodinámicamente menos importante que en la mayoría de los pacientes sin llegar a causar enfermedades valvulares significativas, sin embargo en casos con deformidad valvular extensa y disfuncional requirió sustitución valvular tanto en el SAF primario como secundario. Muchos casos han reportado una alta frecuencia de eventos trombóticos concomitantes con la presencia de anormalidades valvulares54-57.
En un análisis ecocardiográfico retrospectivo realizado por el Grupo de Estudio de ICTUS y SAF, se demostró que el 22% de los pacientes presentó insuficiencia valvular mitral, el 2,8% insuficiencia valvular aórtica, el 9,7% anormalidades en las paredes del ventrículo izquierdo y el 4,2% trombos en el ventrículo izquierdo, por lo que estos resultados sugieren un alto potencial de fuente cardioembólica en los pacientes con SAF58.
Este síndrome puede producir endocarditis pseudo-infecciosa también llamada endocarditis tromboembólica abacteriana, la cual es infrecuente en pacientes con SAF primario, pero no así en los SAF secundarios a LES, en el cual la prevalencia es significativa. Suelen ser de 3-4 mm. La misma se produce por depósitos de anticuerpos anticardiolipina y complemento en el tejido conectivo subendotelial, llevando a la fibrosis y calcificación valvular, sin presencia de microorganismos y no suele acompañarse de reacción inflamatoria11. Los pacientes pueden presentar clínicamente soplos con visualización de vegetación en ecocardiograma, fiebre, hemorragia en astilla, evidencia serológica de actividad lúpica, niveles elevados de anticuerpos antifosfolipídicos y hemocultivos negativos. Todas estas manifestaciones son determinadas por la actividad lúpica.
Presenta una alta tasa de eventos embólicos cercanos al 40% y son más frecuentes a nivel cerebral, presentando eventos isquémicos cerebrovasculares manifestados como ataque isquémicos transitorios (AIT) o accidentes cerebrovasculares (ACV) isquémico.
La diferencia entre la endocarditis pseudo-infecciosa y la infecciosa está dada por tres parámetros:
- Leucocitos bajos.
- Proteína C reactiva aumentada, pero en menor proporción que en la endocarditis infecciosa.
- Anticuerpos antifosfolipídicos en niveles altos59.
La mayor parte de los pacientes presentan insuficiencia mitral leve que rápidamente evoluciona hacia la severidad, la cual exige un recambio valvular. La edad de presentación varía entre los 20-40 años, por lo cual la indicación es el reemplazo valvular con prótesis mecánica, requiriendo anticoagulación. Pero, a pesar del tratamiento anticoagulante, las complicaciones tromboembólicas de esta enfermedad hacen que peligre el normal funcionamiento de la válvula.
Entre los mecanismos patológicos que provocan las anormalidades valvulares se plantea que los AAF mediante el daño valvular pueden promover la formación de trombos, sobre el endotelio valvular lesionado jugando un rol patológico más directo60.
La secuencia de eventos patológicos en la producción de alteraciones valvulares todavía no ha sido identificada. Se plantea a la lesión inmunológica (factiblemente mediada por inmunocomplejos) como posible causa de los depósitos de inmunoglobulinas y de complemento que se hallaron entre los vasos de las paredes del ventrículo izquierdo y en la zona de neovascularización y la zona de la lesión valvular verrugosa según Bidani y colaboradores61 quienes demostraron depósitos de inmunoglobulinas y complemento entre el estroma del endocardio y a lo largo de las valvas y entre las vegetaciones encontradas sobre las valvas de estos pacientes, usando para ellos un marcador específico para anticuerpos anticardiolipinas en los depósitos de inmunoglobulinas61.
Existe evidencia de que los AAF o algunas otras inmunoglobulinas en el suero de pacientes con SAF se adhieren a las células endocárdicas62,63.
Varios efectos biológicos de los AAF se han demostrado in vitro, evidenciándose que pueden incrementar la actividad endotelial, ésta incluye interferencia con la producción y/o liberación de prostaciclina (prostaglandina I), aumentando la producción del factor activación plaquetaria, aumentando la actividad del factor tisular e inhibiendo la liberación del activador del plasminógeno, incrementando el inhibidor del activador del plasminógeno e interfiriendo con el sistema de la proteína C y S del endotelio y la trombomodulina dependiente.
El potencial trombogénico del SAF puede interferir con las funciones de las plaquetas, proteínas plasmáticas y proteínas plasmáticas involucradas en la coagulación de la sangre y la fibrinolisis64,65.
A pesar de que se plantea que la lesión histopatológica en el SAF es la oclusión vascular trombótica sin signos de inflamación66; sin embargo, fueron observados cambios inflamatorios en las válvulas lesionadas de pacientes con SAF primario y secundario por igual57,61,67; además, se ha planteado que no se implican inmunocomplejos en los fenómenos clínicos relacionados con el SAF y por el contrario Pope y colaboradores67 demostraron una disminución total del nivel de complemento con C3 y C4 de pacientes con SAF primarios y enfermedad valvular.
Por otro lado, Tenedios y colaboradores han planteado que el SAF ha sido incriminado en la patogenia de las lesiones cardíacas a partir de investigaciones realizadas con marcadores de células endocárdicas que han demostrado el depósito de AAF que inician el proceso inflamatorio, que recluta complemento hacia la lesión valvular con la aparición de un estado pro inflamatorio, pro adhesivo y pro coagulante a este nivel35,68.
Investigaciones realizadas por Vega-Ostertag y Casper69 demostraron que los AAF incrementan significativamente la transcripción, la función y la expresión del factor tisular, interleukina-8, además éstos disminuyen los AAF, incrementando la expresión de la sintetasa de óxido nítrico, todos a nivel de las células endocárdicas, produciendo daños a nivel de las valvas del endocardio.
A nivel del endocardio, también se presentan las trombosis murales, y en la patogenia de estos se ha demostrado que la estimulación de células pro coagulantes por los AAF, a través de la vía de la B2-GPI, es una de los mecanismos más importantes en el SAF, así como se refiere también a la vía de la MAPK (p38 Mitogen Activited Protein Kinase), quien también juega un papel crucial en la trombosis endocárdica (Figura 3).
La B2-GPI está adherida proteolíticamente al dominio de factor V por la actividad del factor X, dirigida hacia la generación de la B2-PGI como un regulador de la vía intrínseca de la fibrinolisis. La B2-PGI muestra afinidad por los anticuerpos y éstos al unírseles modifican la proteína exponiendo los fosfolípidos de la membrana a los factores de la coagulación incrementando la tendencia trombofílica70 a nivel de todo el endocardio.
Investigaciones realizadas por Miesbach y Mathias han identificado un grupo heterogéneo de anticuerpos contra a trombina en el 20 % de los pacientes estudiados con FAS los anticuerpos fueron el anticuerpo antitrombina IgM y Anticuerpo Antitrombina IgG, estos anticuerpos se relacionaron con la presencia de la B2-PGI en el 96% de los pacientes con SAF y hallaron como resultado que el 67% de los pacientes con anticuerpos antitrombina IgG sufrieron trombosis arteriales71.
Los trabajos realizados por Simoncini y Sapet, han demostrado que los AAF IgG provocan la activación de ROS (Reactive oxygen species), especie de oxígeno reactivo que actúa como segundo mensajero, activando a su vez a la p38 MAPK, y ésta estimula la trascripción del factor tisular, conduciendo finalmente a un incremento de la exposición de moléculas de adhesión vascular endotelial72.
También a nivel del endocardio, se puede afectar el sistema de la conducción. Las madres positivas de SAF con anticuerpos anti-Ro/SSA pueden provocar afecciones en el feto a nivel de sistema de la conducción cardíaco y tener hijo con bloqueos AV congénitos73 .
Compromiso miocárdico
Cardiopatía isquémica
Se presenta en pacientes jóvenes menores de 40 años, representa el 3% de los casos de enfermedad coronaria y es más frecuente encontrar causas distintas a la enfermedad ateromatosa74. En un porcentaje cercano al 3% el IAM es la primera manifestación del SAF.
La presencia de niveles altos de anticuerpos anticardiolipina es un factor de riesgo independiente de IAM5,34,35,74. En el SAF, el 55% de los pacientes presenta TVP y el 25% trombosis de arterias coronarias. El mecanismo exacto de la trombosis en estos pacientes no está dilucidado completamente5. Se asume que el IAM es de causa multifactorial, ya que se ha demostrado que en corazones de ratas con SAF hay aumento de AAF IgG e IgM y disminución de la actividad paraoxonasa del óxido nítrico y aumento del estrés oxidativo con aumento de los radicales libres de oxígeno como el superóxido, peroxinitrito y nitrotiroxina, los cuales aceleran el proceso de aterosclerosis.
Últimamente, se ha considerado al SAF como un factor de riesgo aterosclerótico, ya que se encuentra afectado el endotelio el cual puede generar radicales libres que en presencia de moléculas como el hierro oxidan la molécula de LDL transformándola en LDL oxidada, los ácidos grasos poliinsaturados de la LDL se oxidan y se convierten en lipoperóxidos que a su vez se fragmentan en aldehídos que se combinan covalentemente con la lisina y apolipoproteína B. Al oxidarse la LDL puede activar el factor XII de la coagulación, bloquear la relajación endotelial, contribuir a la lesión del endotelio y activar monocitos. La apolipoproteína B es receptor de macrófagos, los macrófagos activados engloban a las moléculas de LDL oxidada y se transforman en células espumosas que se depositan en el subendotelio y contribuye a la formación de la placa ateromatosa75.
Los AAF al producir estas afecciones podrían incrementar el riesgo de enfermedad coronaria, acelerando el proceso aterosclerótico de causa inmunológica que junto a la tendencia trombofílica pueden conducir al SAF en menores de 45 años76.
Las nuevas investigaciones realizadas por Matsuura Kobayashi77, centra sus investigaciones en el hallazgo de inmunocomplejos conformados por OxiLDL/B2-GPI, es decir inmunocomplejos formados por la B2-PGI (mayor blanco antigénico de los anticuerpos del SAF), con la lipoproteína de bajo peso molecular oxidada, estas dos estructuras conforman inmunocomplejos circulantes que han sido encontrados dentro de la lesión aterosclerótica junto a linfocitos inmunorreactivos, de los pacientes con SAF primario y secundario, además se han visto en el torrente circulatorio de pacientes enfermos de SAF, LES, diabetes mellitus Tipo II e insuficiencia renal crónica (IRC), a favor de esta investigación David y Falco78, plantean que el estrés oxidativo y la peroxidación lipídica en el SAF conducen a una respuesta inflamatoria a bajo grado y crónica que va a incrementar el riesgo cardiovascular y a su vez puede conllevar a la activación del tromboxano plaquetario y a un aumento del riesgo de trombosis arteriales. Por lo cual Vaselini y Alessandri recomiendan el uso del tratamiento antioxidante en la prevención de las trombosis en este síndrome79.
Otros investigadores como Buschmann y Fischer80 demostraron en modelos animales de experimentación que la presencia de AAF activa el complemento C3 y C5, produciendo dos efectos, el primero es el efecto de inducir la trombosis y el segundo es el efecto de activar las células endoteliales.
En investigaciones realizadas en pacientes con IAM y SAF primario, Morel y Gesel han detectado micro partículas pro coagulantes (MP) circulantes, las cuales se han interpretado como marcadores patógenos de incremento de la coagulabilidad, relacionándose con una diversidad de trastornos derivados de la estimulación de células endoteliales, por ejemplo, demostraron que las MP de plaquetas de origen endotelial vascular están muy elevadas en los pacientes con IAM y SAF(de 3 a 6 veces más en los pacientes con IAM y SAF que en el grupo control). Por lo que concluyen que la liberación de micropartículas puede estar contribuyendo al desarrollo de trombos intracardíacos e intracoronarios81.
Existen otros estudios en pacientes con síndrome coronario agudo y SAF hechos por Veres y Lakos82 donde demostraron que los anticuerpos ANTI B2-PGI IgA estuvieron significativamente más elevados en la angina inestable y en el IAM con elevación del ST y SAF que en los síndromes coronarios agudos sin SAF; además, se demostró mayor frecuencia de elevación de los anticuerpos anti B2-GPI isotipo IgA en los hombres que en las mujeres y mucho más frecuente en los jóvenes que en los viejos.
Hipertensión pulmonar
El compromiso pulmonar en el SAF puede ser diverso produciendo embolia pulmonar, hipertensión pulmonar, trombosis arterial pulmonar. La presencia de hipertensión pulmonar en el SAF es de 1,5-5%, los anticuerpos actúan en la génesis de la hipertensión pulmonar ya que se produce por trombosis in situ o por alteración del equilibrio tromboxano/prostaciclina o por daño directo sobre el endotelio lo cual podría dar lugar a fenómenos de remodelación endotelial y favorecimiento de la síntesis de sustancias potentes vasoconstrictoras como la endotelina I y es por eso que también pueden producir hipertensión pulmonar sin tromboembolia83.
Conclusión
El síndrome antifosfolipídico o síndrome de Hughes presenta un manejo multidisciplinario, aunque poco se conoce, la afectación cardiovascular es común en este síndrome. Se ha demostrado que su asociación con el lupus eritematoso sistémico en forma de SAF secundario debe obligar a realizar una búsqueda exhaustiva de patología cardiovascular. Actualmente, el SAF debe ser considerado como un factor de riesgo cardiovascular, ya que puede interferir con la función endotelial, aunque los datos sobre este punto son hasta el momento insuficientes.
Recursos financieros
El autor no tuvo ningún apoyo económico para la investigación.
Conflicto de intereses
El autor declara no tener conflicto de intereses.
1. Hughes GRV. Thrombosis, abortion, cerebral disease, and the lupus anticoagulant. Br Med J 1983;287:1088-9. [ Links ]
2. Sáez RA, Pons AG, Wainstein EG, Germain AAM. Síndrome de Anticuerpos Antifosfolípidos. Rev Med Clin Condes 2007;18(4):376-382. [ Links ]
3. Contreras MAR. Inmunopatogenia del Síndrome Antifosfolípido. Rev Chil Reumatol 2009; 25(4):149-155. [ Links ]
4. Gómez Moreno R, Monge Ropero N, Calvo Cebrián A, Fraga Campo S. Síndrome Antifosfolípido. Med Gen 2004;62:157-163. [ Links ]
5. Uribe CE, Cárdenas JM, Cabrales J, Bohórquez R, Roa NL, Beltrán J, Urina M. Infarto agudo del miocardio como primera manifestación del síndrome antifosfolípido primario en un paciente de veinticuatro años. Rev Colomb Cardiol 2005;12:135-139. [ Links ]
6. Wassermann A, Neisser A, Bruck C. Eine serodiagnostiche Reaktion bei Syphilis. Deutsche Med Wochenschr 1906;32:745-6. [ Links ]
7. Pangborn MC. A new serologically active phospholipid from beefbei Syphilis. Deutsche Med Wochenschr 1906;32:745-6. [ Links ]
8. Hughes GRV. The antiphospholipid syndrome. Lancet 1993;342: 341-4. [ Links ]
9. Hughes GRV. The antiphospholipid syndrome. Lupus 1996;5: 345-6. [ Links ]
10. Harris EN, Gharavi AE, Boey ML, et al. Anticardiolipin antibodies: detection by radioimmunoassay and association with thrombosis in systemic lupus erythematosus. Lancet 1983;2:1211-4. [ Links ]
11. Meroni PL, Del Papa N, Raschi E, et al. b2-Glycoprotein I as a 'cofactor' for anti-phospholipid reactivity with endothelial cells. Lupus 1998;7 (Suppl 2):S44-S47. [ Links ]
12. García-García C. Anticuerpos antifosfolípido y síndrome antifosfolípido: actitudes diagnósticas y terapéuticas. Actas Dermosifiliogr 2007;98:16-23. [ Links ]
13. Koniari I, Siminelakis SN, Baikoussis NG, Papadopoulos G, Goudevenos J, Apostolakis E. Antiphospholipid syndrome; its implication in cardiovascular diseases: a review. J Cardiothorac Surg 2010;5:101. [ Links ]
14. Galarza-Maldonado C, Cervera Segura R, Urgilez Morejón H. Síndrome antifosfolipídico: veintiún años después. Rev Colomb Reumatol 2004;11(1):48-54. [ Links ]
15. Wilson WA, Gharavi AE, Kuikets T, et al. International consensus statement on preliminary classification criteria for definite Antiphospholipid syndrome. Report of an international workshop. Arthritis Rheum 1999;42:1309-1311. [ Links ]
16. Alarcón-Segovia D, Pérez-Vázquez ME, Villa AR, Drenkard C, Cabiedes J. Preliminary classification criteria for the antiphospholipid syndrome within systemic lupus erythematosus. Semin Arthritis Rheum 1992;21:275-86. [ Links ]
17. Alfaro Pacheco R. Síndrome Antifosfolípido. Rev Med De Costa Rica y Centro América 2009;LXVI (589): 313-317. [ Links ]
18. Young M. Antiphospholipid syndrome: multiple mechanisms. Clin Exp Immunol 2004;136:393-401. [ Links ]
19. Espinosa G, Cervera R. Antiphospholipid syndrome. Arthritis Res Ther 2008;10:230-238. [ Links ]
20. Salmon J, Groot P. Pathogenic role of antiphospholipid antibodies. Lupus 2008;17:406-411. [ Links ]
21. Myones BL, Mccurdy D. The Antiphospholipid syndrome: immunological and clinical aspect. Clinical spectrum and treatment. J Rheumatol 2000;27:20-28. [ Links ]
22. Inanc M, Radway-Bright EL, Isenberg DA. B2 glicoprotein I and anti B2 glicoprotein antibody. Br J Rheumatol 1997;36:1247-1257. [ Links ]
23. Petri M. Treatment of lupus. Cap 8. En: Clinical symposium. Special lectures. Waschington. ACR. Syllabus.1997. [ Links ]
24. Cabral AR, Alarcon-Segovia D. Autoantibodies in SLE. Curr Opin Rheumatol 1997;9:387-392. [ Links ]
25. Shrivastava A, Dwivedi S, Aggarwal A, et al. Anticardiolipin and antiB2 glicoprotein antibodies in patients with SLE. Association with the presence of seizures. Lupus 2001;10:45-50. [ Links ]
26. Rand JH, Xuan-Wu X, Andree H, et al. Antiphospholipid antibodies accelerate plasma coagulation by inhibiting annexin-V binding phospholipid: a lupus procoagulant phenomenon. Blood 1998;92:1652-1660. [ Links ]
27. Meng Ch, Lockshim M. Pregnancy in Lupus. Curr Opin Rheumatol 1999;11:348-351. [ Links ]
28. Rand JH. Antiphospholipid antibody syndrome. New insight on the thrombosis mechanism. Am J Med Sci 1998;16:142-151. [ Links ]
29. Stone S, Khamastha MA, Poston L. Placentation, antiphospholipid syndrome and pregnancy outcome. Lupus 2001;10:67-74. [ Links ]
30. Gharavi AE, Pierangeli SS, Levy RA, et al. Mechanism of pregnancy loss in antiphospholipid syndrome. Clin Obstet Gynecol 2001;44:11-19. [ Links ]
31. Rand JH, Xuan-Wu X, Andree H, et al. Pregnancy loss in the Antiphospholipid syndrome. A possible thrombogenic mechanism. N Engl J Med 1997;337:154-160. [ Links ]
32. Bertolaccini ML, Atsumi T, Lanchbury JT, et al. Plasma tumor necrosis factor alfa levels and the 238. A promoter polymorphism in patients with Antiphospholipid syndrome. Thromb Haemost 2001;85:198-203. [ Links ]
33. Olvestad A, Kanestrom A, Tegner P, et al. Anticardiolipin autoantibodies and pulmonary embolism. Scand J Rheumatol 2000;29:330-332. [ Links ]
34. Gómez Puerta JA, López F, Molina JF. Manifestaciones cardíacas de las enfermedades reumáticas. Rev Colomb Reumatol 2002;9(3):203-213. [ Links ]
35. Tenedios F, Erkan D, Lockshin MD. Cardiac Manifestations in the Antiphospholipid Syndrome. Rheumatic Diseases Clinics of North America 2006;32(3):491-507. [ Links ]
36. Khamashta MA, Cervera R, Asherson RA, Font J, Gil A, Coltart DJ, Vazquez JJ, Pare C, Ingelmo M, Oliver J, Hughes GRV. Association of antibodies against phospholipids with heart valve disease in systemic lupus erythematosus. Lancet 1990;335:1541-1544. [ Links ]
37. Nihoyannopoulos P, Gomez PM, Joshi J, Loizou S, Walport MJ, Oakley CM. Cardiac abnormalities in systemic lupus erythematosus: association with raised anticardiolipin antibodies. Circulation 1990;82:369-375. [ Links ]
38. Cervera R, Font J, Pare C, Azqueta M, Perez-Villa F, Lopez-Soto A, Ingelmo M. Cardiac disease in systemic lupus erythematosus: prospective study of 70 patients. Ann Rheum Dis 1992;51:156-159. [ Links ]
39. Roldan CA, Shively BK, Lau CC, Gurule FT, Smith EA, Crawford MH. Systemic lupus erythematosus valve disease by transesophageal echocardiography and the role of antiphospholipid antibodies. J Am Coll Cardiol 1992;20:1127-1134. [ Links ]
40. Gleason CB, Stoddard MF, Wagner SG, Longaker RA, Pierangeli S, Harris EN. A comparison of cardiac valvular involvement in the primary antiphospholipid syndrome versus anticardiolipin-negative systemic lupus erythematosus. Am Heart J 1993;125:1123-1129. [ Links ]
41. Ford SE, Lillicrap D, Brunet D, Ford P. Thrombotic endocarditis and lupus anticoagulant. Arch Pathol Lab Med 1989;113:350-353. [ Links ]
42. Libman E, Sacks B. A hitherto undescribed form of valvular and mural endocarditis. Arch Intern Med 1924;33:701-737. [ Links ]
43. Baehr G, Klemperer K, Schifrin A. A diffuse disease of the peripheral circulation usually associated with lupus erythematosus and endocarditis. Trans Assoc Am Physicians 1935;50:139-155. [ Links ]
44. Gross L. The cardiac lesion in Libman-Sacks disease with a consideration of its relationship to acute diffuse lupus erythematosus. Am J Pathol 1940;16:375-408. [ Links ]
45. Shearn MA. The heart in systemic lupus erythematosus: a review. Am Heart J 1959;58:452-466. [ Links ]
46. Bridgen W, Bywaters EG, Lessof MH, Ross IP. The heart in systemic lupus erythematosus. Br Heart J 1960;22:1-16. [ Links ]
47. Kong TQ, Kellum RE, Haserick JR. Clinical diagnosis of cardiac involvement in systemic lupus erythematosus: a correlation of clinical and autopsy findings in thirty patients. Circulation 1962;26:7-11. [ Links ]
48. Bulkley BH, Roberts WC. The heart in systemic lupus erythematosus and the changes induced in it by corticosteroid therapy: a study of 36 necropsy patients. Am J Med 1975;58:243-264. [ Links ]
49. Ansari A, Larson PH, Bates HD. Cardiovascular manifestations of systemic lupus erythematosus: current perspective. Prog Cardiovasc Dis 1985;27:421-434. [ Links ]
50. Mandell BF. Cardiovascular involvement in systemic lupus erythematosus. Semin Arthritis Rheum 1987;17:126-141. [ Links ]
51. Vianna JL, Khamashta MA, Ordi-Ros J, Font J, Cervera R, Lopez-Soto A, Tolosa C, Franz J, Selva A, Ingelmo M, Vilardell M, Hughes GRV. Comparison of the primary and secondary antiphospholipid syndrome: a European multicenter study of 114 patients. Am J Med 1994;96:3-9. [ Links ]
52. Leung WH, Wong KL, Lau CP, Wong CK, Liu HW. Association between antiphospholipid antibodies and cardiac abnormalities in patients with systemic lupus erythematosus. Am J Med 1990;89:411-419. [ Links ]
53. Giunta A, Picillo U, Maione S, Migliaresi S, Valentini G, Arnese M, Losardo L, Marone G, Tirri G, Condorelli M. Spectrum of cardiac involvement in systemic lupus erythematosus: echocardiographic, echo-Doppler observations and immunological investigation. Acta Cardiol 1993;48:183-197. [ Links ]
54. Barbut D, Borer JS, Gharavi A, Wallerson D, Devereux RB, Supino P, Suite NDA. Prevalence of anticardiolipin antibody in isolated mitral or aortic regurgitation, or both, and possible relation to cerebral ischemic events. Am J Cardiol 1992;70:901-905. [ Links ]
55. Alvarez-Blanco A, Egurbide-Arberas MV, Aguirre-Errasti C. Severe valvular heart disease in a patient with primary antiphospholipid syndrome. Lupus 1994;3:433-434. [ Links ]
56. Nickele GA, Foster PA, Kenny D. Primary antiphospholipid syndrome and mitral valve thrombosis. Am Heart J 1994;128:1245-1247. [ Links ]
57. Jafar MZ, Chester MM, Gorcsan J. Transesophageal echocardiographic detection of multiple mitral valve masses in primary antiphospholipid syndrome with stroke. Am Heart J 1994;127:445-446. [ Links ]
58. Jurado MO, Durán J, Martínez A, Castellón JM, Gutiérrez MA. Infarto agudo al miocardio en un hombre joven sin ateromatosis coronaria, como forma de presentación de síndrome antifosfolípido primario. Rev Méd Chile 2009;137:1478-1481. [ Links ]
59. Asherson RA, Cervera R. Antiphospholipid antibodies and the heart. Lessons and pitfalls for the cardiologist. Circulation 1991;84:920-923. [ Links ]
60. Asherson RA, Tikly M, Staub H, Wilmshurst, et al. Infective endocarditis, rheumatoind factor, and anticardiolipin antibodies. Ann Reu Dis 1990;49: 107-108. [ Links ]
61. Ziporen L, Goldberg I, Kopolovic Y, Arad M, Sandbank Y, Ordi-Rose J, Vilardell-Tarress M, De Torres I, Adler Y, Weinberger A, Asherson RA, Shoenfeld Y. Libman Sacks endocarditis: the possible pathogenic role of anti-cardiolipin antibodies deposited at the valve subendothelium. Lupus 1995;4(Suppl 2):100. [ Links ]
62. McGrae KR, de Michelle A, Samuels P, Roth D, Kuo A, Meg QH, Rauch J, Cines DA. Detection of endothelial cell reactive immunoglobulin in patients with antiphospholipid antibodies. Br J Haematol 1991;79:595-605. [ Links ]
63. Cervera R, Khamashta MA, Font J, Ramirez J, Cruz D, Montalban J, Lopez-Soto A, Asherson RA, Ingelmo M, Hughes GRVH. Antiendothelial cell antibodies in patients with the antiphospholipid syndrome. Autoimmunity 1991;11:1-6. [ Links ]
64. Harris EN. Antiphospholipid antibodies. Br J Haematol 1990;74:1-9. [ Links ]
65. Reyes H, Dearing L, Shoenfeld Y, Peter JB. Antiphospholipid antibodies: a critique of their heterogeneity and hegemony. Semin Thromb Hemost 1994;20:89-100. [ Links ]
66. Ford SE, Kennedy L, Ford PM. Clinicopathologic correlations of antiphospholipid antibodies: an autopsy study. Arch Pathol Lab Med 1994;118:491-495. [ Links ]
67. Pope JM, Canny CLB, Bell DA. Cerebral ischemic events associated with endocarditis, retinal vascular disease, and lupus anticoagulant. Am J Med 1991;90:299-309. [ Links ]
68. Tenedios F, Erkan D, Lockshin MD. Cardiac involvement in the antiphospholipid syndrome. Lupus 2005;14(9):691-6. [ Links ]
69. Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum 2005;52(5):1545-54. [ Links ]
70. Atsumi T, Amengual O, Yasuda S, Matsuura E, Koike T. Research around beta2-glycoprotein I: A major target for antiphospholipid antibodies. Autoimmunity 2005;38(5):377-81. [ Links ]
71. Miesbach W, Matthias T, Scharrer I. Identification of thrombin antibodies in patients with antiphospholipid syndrome. Ann N Y Acad Sci 2005;1050:250-6. [ Links ]
72. Simoncini S, Sapet C, Camoin-Jau L, Bardin N, Harle JR, Sampol J, Dignat-George F, Anfosso F. Role of reactive oxygen species and p38 MAPK in the induction of the pro-adhesive endothelial state mediated by IgG from patients with anti-phospholipid syndrome. Int Immunol 2005;17(4):489-500. [ Links ]
73. Tincani A, Biasini-Rebaioli C, Cattaneo R, Riboldi P. Nonorgan specific autoantibodies and heart damage. Lupus 2005;14(9):656-9. [ Links ]
74. Sevilla B, Roldán I, Baello P, Mora V, Salim M, Salvador A. Síndrome antifosfolípido con infarto de miocardio y afectación valvular aórtica. Rev Esp Cardiol 2000;53:1534-1536. [ Links ]
75. Gómez Padrón MV, Torres W, Gómez Padrón EI, Mérida Álvarez O. ¿Debería ser considerado el síndrome antifosfolípido un nuevo factor de riesgo cardiovascular? Rev Cubana Med 2008;47(1). [ Links ]
76. Delgado Alves J, Mason LJ. Ames P, Chen P, Rauch J, Levine JS. Antiphospholipid antibodies are associated with enhanced oxidative stress, decreased plasma nitric oxide and paraoxonase activity in an experimental mouse model. Rheumatology 2005;44(10):1238-1244. [ Links ]
77. Matsuura E, Kobayashi K, Inoue K, Lopez LR, Shoenfeld Y. Oxidized LDL/![]() 2-glycoprotein I complexes: new aspects in atherosclerosis. Lupus 2005;14 (9): 736-741. [ Links ]
2-glycoprotein I complexes: new aspects in atherosclerosis. Lupus 2005;14 (9): 736-741. [ Links ]
78. Davi G, Falco A. Oxidant stress, inflammation and atherogenesis. Lupus 2005;14(9):760-764. [ Links ]
79. Valesini G, Alessandri C. New facet of antiphospholipid antibodies. Ann N Y Acad Sci 2005;1051:487-97. [ Links ]
80. Buschmann C, Fischer C, Ochsenhirt V, Neukirch C, Lackner KJ, von Landenberg P. Generation and Characterization of Three Monoclonal IgM Antiphospholipid Antibodies Recognizing Different Phospholipid Antigens. Ann N Y Acad Sci 2005;1051:240-54. [ Links ]
81. Morel O, Jesel L, Freyssinet JM, Toti F. Elevated levels of procoagulant microparticles in a patient with myocardial infarction, antiphospholipid antibodies and multifocal cardiac thrombosis. Thromb J 2005;3(1):15. [ Links ]
82. Veres K, Lakos G, Kerenyi A, Szekanecz Z, Szegedi G, Shoenfeld Y, Soltesz P. Antiphospholipid antibodies in acute coronary syndrome. Lupus 2004;13(6):423-7. [ Links ]
83. Triana Moreno LC. Compromiso pulmonar en el síndrome antifosfolípido. Rev Colomb Neumonol 2004;16(4);236-240. [ Links ]

