










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkInsuficiencia cardíaca
On-line version ISSN 1852-3862
Insuf. card. vol.9 no.2 Ciudad Autónoma de Buenos Aires June 2014
INSUFICIENCIA CARDÍACA DESDE SUS ORÍGENES A LA ACTUALIDAD
En búsqueda del Santo Grial
Capítulo dos
Sergio V. Perrone1
1 Médico Cardiólogo. Insuficiencia cardíaca. Hipertensión pulmonar. Trasplante cardíaco. Instituto FLENI. Ciudad de Buenos Aires. República Argentina.
Instituto Argentino de Diagnóstico y Tratamiento. Ciudad de Buenos Aires. República Argentina.
Hospital Alta Complejidad en Red El Cruce "Dr. Néstor Carlos Kirchner". Florencio Varela. Buenos Aires. República Argentina.
Instituto Cardiovascular Lezica. San Isidro. Buenos Aires. República Argentina.
Hospital Italiano de Mendoza. Mendoza. República Argentina.
Correspondencia: Dr. Sergio V. Perrone.
Montañeses 2325. CP: C1428AQK. Ciudad Autónoma de Buenos Aires. República Argentina.
E-mail: svperrone@netizen.com.ar
Tel: (54-11) 5777-3200 - Fax: (54- 11) 5777-3209
Recibido: 25/01/2014
Aceptado: 03/04/2014
Resumen
La presente revisión es una recopilación de la Historia de la Medicina en la Insuficiencia Cardíaca en cuatro capítulos que serán presentados durante el año en curso. Se describirán los esfuerzos de la medicina y ciencias afines para combatir este grave problema que es el punto final de casi todas las patologías cardiovasculares. Nuestro objetivo es tratar de correlacionar en este documento la historia de los diferentes modelos fisiopatológicos y terapéuticos de la insuficiencia cardíaca y su evolución hasta la actualidad, de una manera inseparable de la Historia de la Medicina y, sobre todo de la Historia de la Cardiología.
Palabras clave: Historia de la Medicina ; Insuficiencia cardíaca ; Revisión ; Historia de la Cardiología
Summary
In search of the Holy Grail
This review is a compilation of the History of Medicine in Heart Failure that we will present in four chapters during the current year. They will describe the efforts of medicine and related sciences to combat this serious problem which is the end point of almost all cardiovascular pathologies. Our objective is to try to correlate in this document the history of different pathophysiological and therapeutic models of heart failure and its evolution to the present in an inseparable way to the History of Medicine and, particularly, to the History of Cardiology.
Keywords: History of Medicine ; Heart failure ; Review ; History of Cardiology
Resumo
Em busca do Santo Graal
Esta revisão é uma compilação da História da Medicina na Insuficiência Cardíaca em quatro capítulos que serão apresentados durante o ano em curso. São descritos os esforços das ciências médicas e afins para combater este grave problema que é o ponto final de quase todas as doenças cardiovasculares. Nosso objetivo é tentar correlacionar neste documento a história dos modelos fisiopatológicos e terapêuticos da insuficiência cardíaca e sua evolução até o presente, de forma inseparável da História da Medicina, especialmente a História da Cardiologia.
Palavras-chave: História da Medicina ; Insuficiência cardíaca ; Revisão ; História da Cardiologia
Fisiopatología de la insuficiencia cardíaca
La curiosidad del hombre, la necesidad de detectar las causas que provocaban la insuficiencia cardíaca y su progresión, la necesidad de encontrar terapéuticas cada vez más efectivas y seguras, determinaron, a través de los años, la evolución de los modelos fisiopatológicos de la insuficiencia cardíaca. Para resumir su evolución, podríamos considerar a la insuficiencia cardíaca como un estado fisiopatológico caracterizado por la incapacidad del corazón mantener un volumen minuto (VM) adecuado que permita abastecer el metabolismo de los tejidos1-13.
El deterioro de la función miocárdica desarrolla una serie de complejos mecanismos compensadores que deteminan el síndrome de insuficiencia cardíaca y que dan como resultado la presencia de signos y síntomas de congestión pulmonar y/o sistémica y de bajo gasto cardíaco1-4,6-9,12,14-16.
El daño cardíaco primario por una noxa, una sobrecarga hemodinámica excesiva sobre el ventrículo, o ambas, desencadenan esta serie de mecanismos compensadores que tienden a mantener un gasto cardíaco adecuado que permita una perfusión de los
diferentes órganos tanto en reposo como en la actividad física. El agotamiento de estos recursos compensadores determina la aparición de signos y síntomas de insuficiencia cardíaca que culminan en un progresivo deterioro la clase funcional, la calidad de vida y, finalmente, la supervivencia de los pacientes4,5,7,14,17-20 (Figura 1).
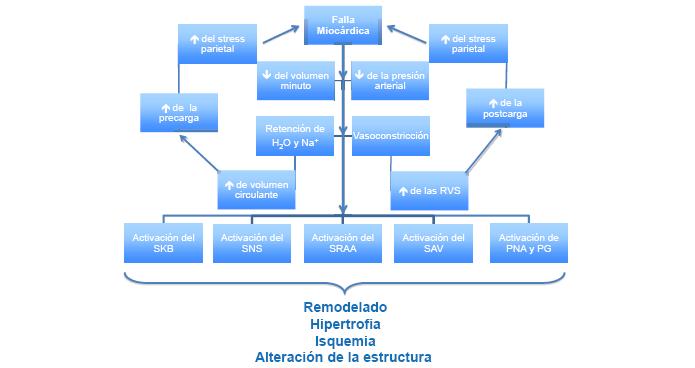
Figura 1. Mecanismos de adaptación de la falla cardíaca.
RVS: resistencias vasculares sistémicas. SKV: sistema kalicreína-bradicinina. SNS: sistema nervioso simpático. SRAA: sistema renina angiotensina aldosterona. SAV: sistema arginina vasopresina. PNA: péptido natriurético atrial. PG: prostaglandinas.
En el esfuerzo por compensar el deterioro miocárdico intervienen diversos mecanismos compensadores18,21, que intentan mantener un adecuado gasto cardíaco, muchas veces insuficientes y, ante su agotameinto, terminan por desencadenar los signos y síntomas que llevan al diagnóstico clínico de insuficiencia cardíaca22.
En la actualidad, existen algunos biomarcadores que nos permiten detectar la activación de los mencionados mecanismos independientemente de la presencia o no de signosintomatología de insuficiencia cardíaca, lo cual permite su detección más precoz e incluso determinar pronósticos de los pacientes portadores de falla cardíaca23-33.
El principio de una nueva era
La función ventricular y, en general, la función cardíaca global, depende de la interacción de factores que regulan el volumen de sangre expulsado por el corazón (VM): precarga, postcarga y contractilidad miocárdica16,34,35. Estos factores modifican el volumen que el corazón expulsa en cada latido. El cuarto factor determinante es la frecuencia cardíaca, la cual, variando el número de latidos por minuto influye directamente en el volumen cardíaco eyectado por minuto36,37.
La disminución del volumen sistólico determinado por la afección cardíaca conlleva un incremento del volumen cardíaco residual, provocando un incremento del volumen de fin de diástole y, consecuentemente, de las presiones intracardíacas38-40.
Esta disminución del volumen eyectado desencadena la activación de mecanismos que, al detectar una caída del volumen circulante, promueven la retención de agua y sodio incrementando la precarga cardíaca41-43. El problema se agrava al encontrar que ese corazón deteriorado no es capaz de manejar ese aporte extra de volumen circulante. Por otra parte, la caída del volumen eyectado se acompaña inicialmente de una caída de las cifras de tensión arterial que inducen un incremento de la vasconstricción con la finalidad de perfundir adecuadamente órganos nobles. Esta vasoconstricción incrementa las resistencias vasculares sistémicas aumentando la postcarga cardíaca que, como es de suponer, el corazón dañado no puede manejar adecuadamente, conformando así un círculo vicioso38.
Estos incrementos de pre y postcarga, acompañados de un incremento de la frecuencia cardíaca tienen como finalidad el intento de mantener un adecuado VM, pero finalmente terminan por requerir del músculo cardíaco dañado un esfuerzo extra, con mayor consumo de oxígeno miocárdico, el cual, ante la imposibilidad de realizarlo, se traducirá en signos y síntomas de congestión y bajo gasto cardíaco.
Estos mecanismos intrínsecos de la regulación de la función cardíaca están acompañados por mecanismos neurohumorales entre los que se destaca la importancia del sistema nervioso simpático y la producción de sustancias vasoactivas en el que desempeñan un papel de máxima importancia el riñón y la médula suprarrenal38,44-48.
Mecanismo de Frank y Starling
El aumento de la precarga, determinado por la caída del volumen eyectado y la retención de agua y sodio, lleva a la utilización del llamado mecanismo de Frank y Starling (Otto Frank 1865-1944 (Figura 2) y Ernest Starling 1866-1927 (Figura 3) como uno de los primeros mecanismos intrínsecos del músculo cardíaco por compensar la falla cardíaca16,41.

Figura 2. Otto Frank (1865-1944).

Figura 3. Ernest Starling (1866-1927).
Según la ley postulada por Frank y Starling, la energía liberada en cada contracción es proporcional al estiramiento de la fibra muscular cardíaca durante la diástole y por tanto depende fundamentalmente del volumen telediastólico38,49-51.
Así, el incremento del volumen y de la presión de fin de diástole provocan un incremento de la energía liberada en cada contracción que es proporcional a la elongación de la fibra muscular. Por lo tanto, un aumento de la precarga conduce a un alargamiento de los sarcómeros con superposición de los miofilamentos gruesos y delgados, incrementando la fuerza contracción y el trabajo cardíaco (Figura 4).
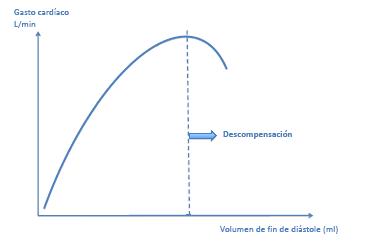
Figura 4. Mecanismo de Frank y Starling. Un aumento en el volumen de fi n de diástole, lleva a un mayor estiramiento de la fi bra muscular con el consecuente incremento de la fuerza de contracción e incremento del volumen eyectado (mecanismo de Frank y Starling) hasta un punto en que el estiramiento de la fi bra muscular deja de producir un incremento en la fuerza de contracción desencadenando el fenómeno de descompensación.
El daño miocárdico es el determinante de la alteración de la capacidad de eyección del ventrículo, incrementando el volumen de sangre al final de la diástole con el consecuente aumento de la tensión de las fibras miocárdicas. El ventrículo responde a ese incremento de la tensión diastólica o precarga incrementando su fuerza de contracción que, junto con el incremento del volumen de fin de diástole contribuirán en su intención de mantener un volumen de eyección adecuado en cada latido. Pero, en el corazón insuficiente, aumentos importantes del volumen de llenado se correlacionan paulatinamente con incrementos menores del volumen de eyección, hasta llegar a un límite, en que el músculo cardíaco claudica, y aparecen signos de congestión vascular y el volumen de eyección comienza a disminuir determinando la falla anterógrada52,53.
Este mecanismo compensador es un importante precursor de la activación neurohumoral y su utilidad en la mantención de una adecuada perfusión de los órganos es limitada.
Los mecanismos compensadores neurohormonales actúan, en parte, sinérgicamente, aumentando la precarga (al retener agua y sodio) para aprovechar al máximo el mecanismo de Frank y Starling; pero, por otro lado, incrementando la postcarga (al provocar vasoconstricción e incremento de las resistencias vasculares) para intentar mejorar la perfusion de los órganos nobles; pero, imponiendo al corazón dañado, un esfuerzo extra54.
A esta altura del daño cardíaco, su accionar es altamente dependiente de la precarga y de la postcarga, determinando que pequeños cambios se correlacionen con significativas modificaciones del gasto cardíaco55 (Figuras 5 y 6).

Figura 5. Un aumento en el volumen inicial del ventrículo (un aumento de la precarga), lleva a un aumento del volumen eyectivo por el mecanismo de Frank y Starling.

Figura 6. El aumento de la postcarga ofrece una mayor resistencia al vaciamiento ventricular, aumentando el volumen residual. Si el retorno venoso no varía, ocurre un desplazamiento de la gráfi ca hacia la derecha (de A a B).
El daño miocárdico promueve fibrosis y la distensión crónica de la fibra miocárdica promueve hipertrofia y en conjunto determinan el remodelado de la pared ventricular. El incremento de los volúmenes y de la presión de fin de diástole y de sístole provocado por los mecanismos compensadores activados, incrementa los requerimientos metabólicos, favoreciendo la isquémia miocárdica, fundamentalmente a nivel subendocárdico, contribuyendo a una mayor insuficiencia56.
Esta activación de mecanismos compensadores, la hipertrofia miocárdica y el remodelado cardíaco y vascular llevarán a la generación de un círculo vicioso en los mecanismos fisiopatológicos de la insuficiencia cardíaca y su consecuente progresión.
Activación del sistema neurohumoral
La activación del sistema neurohumoral es un mecanismo de defensa necesario para la compensación de la caída en la actividad contráctil del músculo cardíaco57-61 (Figura 7). Ésta se produce a expensas de la interacción de diferentes mecanismos que incluyen al sistema simpático, la médula suprarrenal, el sistema renina-angiotensina-aldosterona (SRAA)62 y los sistemas humorales, arginina-vasopresina (SAV)63,64, calicreína-bradicinina (SKB)65-70, prostaglandinas (PG)70,71 y péptido natriurético atrial (PNA)46,62,72-74 (Figura 1).

Figura 7. Sistema neurohumoral. Activación de los mecanismos compensadores. SNS: sistema nervioso simpático. SRAA: sistema renina, angiotensina, aldosterona. FC: frecuencia cardíaca. RVS: resistencias vasculares sistémicas. ADH: hormona antidiurética (sistema arginina-vasopresina).
En pacientes con insuficiencia cardíaca, el flujo sanguíneo cardíaco y cerebral se mantiene a expensas de una redistribución vascular, por medio de la cual se logra una reducción del flujo esplácnico, hepático y renal. Esta redistribución es consecuencia de mecanismos reguladores ejercidos por el sistema simpático, el SRAA, el SAV, la síntesis de prostaglandinas, y de péptido atrial natriurético. También a nivel de la musculatura esquelética se produce una importante reducción del flujo sanguíneo, lo que determina una disminución de la capacidad de ejecicio75.
Cuando la activación de estos mecanismos neurohumorales logra su objetivo, el cuadro de insuficiencia cardíaca logra un equilibrio de compensación y la signosintomatología resulta nula o escasa. Pero, cuando esta activación no logra mantener dicha estabilidad, determinará una sobreactivación del sistema que terminará resultando, en el largo plazo, perjudicial47,76,77. Esta incapacidad de mantener la estabilidad puede deberse a varias causa entre las que podemos mencionar el agravamiento de la falla miocárdica o una mayor exigencia al músculo cardíaco, debido a alguna intercurrencia, alguna transgresión (dietética o medicamentosa), o a una actividad física o incluso psíquica desmedida.La activación del sistema neurohumoral ha sido atribuida, fundamentalmente, a la caída de la perfusión, secundaria a la caída del gasto cardíaco78,79.
La activación de estos mecanismos neurohumorales determinan, entre otros, la re-expansión de volumen sanguíneo circulante en la intención de mantener una adecuada perfusión de los órganos vitales80-83, pero sus efectos secundarios, suelen profundizar o perpetuar la falla cardíaca miocárdica en un círculo vicioso:
- Retención progresiva de agua y sodio incrementando el volumen circulante.
- Vasoconstricción incrementando la postcarga.
- Hipertensión venocapilar.
- Congestión pulmonar y sistémica.
- Arritmias.
Sistema nervioso simpático
La primera respuesta al daño miocárdico se refleja en una caída del volumen sistólico con la consecuente caída de la tensión arterial. Esta caída de la tensión arterial es interpretada como una caída del volumen circulante, produciendo una activación del sistema nervioso simpático. La activación de este sistema determina la liberación de catecolaminas a la circulación general (fundamentalmente de noradrenalina) con la finalidad de incrementar la fuerza contráctil, aumentar la frecuencia cardíaca y provocar vasoconstricción84-87 (Figura 8). Este incremento de las catecolaminas circulantes promueve una disminución de los depósitos de la misma y un down-regulation de los receptores beta-adrenérgicos con la consecuente y progresiva reducción de la respuesta a las mismas88-90.
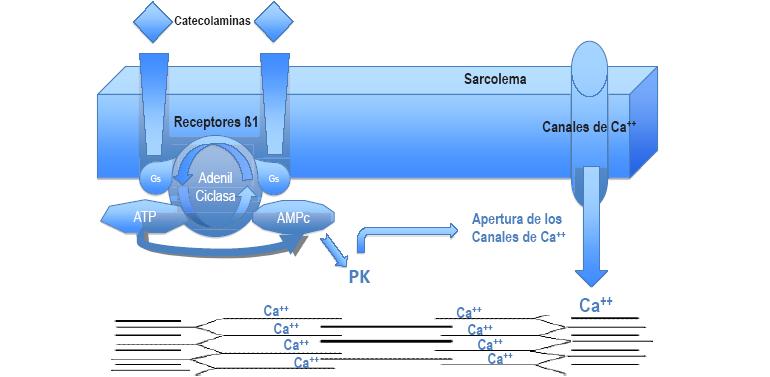
Figura 8. Efecto de las catecolaminas a nivel miocárdico. Las catecolaminas circulantes estimulan los receptores que activan la proteínas de membrana Gs activando la adenilciclasa que transforma el ATP en AMP cíclico, cuyo incremento activa la proteincinasa AMPc dependiente (PK) que facilita la fosforilación de proteínas intracelulares que provocan la apertura de los canales de calcio incrementando la disponibilidad de calcio intracelular a las miofi brillas y, consecuentemente, aumentando la contractilidad miocárdica.
Esta estimulación excesiva de los receptores adrenérgicos produce un incremento de las resistencias vasculares sistémicas, y el consecuente incremento de las presiones de llenado de las cavidades cardíacas e hipertrofia miocárdica.
Los cambios producidos en el músculo cardíaco determinan cambios en el fenómeno relajación/contracción, con el desarrollo de hipertrofia/dilatación de cavidades cardíacas que determinan alteraciones en la relación aporte/consumo de oxígeno miocárdico, prolongación de los potenciales de acción y las alteraciones electrofisiológicas como trastornos de conducción y arritmias91.
Los niveles elevados de catecolaminas constituyen un determinante de mal pronóstico que refleja el grado de activación del sistema nervioso simpático, aunque la relación niveles de catecolaminas/mortalidad no sea lineal87,92,93.
Se postularon diferentes mecanismos que justifican el papel pronóstico que poseen los niveles de catecolaminas (noradrenalina) en pacientes portadores de insuficiencia cardíaca94.
A las catecolaminas se atribuye, además, un efecto tóxico directo sobre el miocardio al incrementar los niveles de Ca++ intracelular, determinando hipercontracción, apoptosis y necrosis miocárdica y/o exacerbación de las arritmias cardíacas95-98. También, como comentamos, se relacionaron los niveles elevados de las catecolaminas con una disminución (down-regulation) de los receptores beta-adrenérgicos miocárdicos (disminución de los receptores beta1 y desacople de la señal de los receptores beta2), y con el incremento de lasresistencias vasculares sistémicas consecuencia de la vasoconstricción periférica, condicionando la necesidad de un mayor esfuerzo miocárdico y un mayor deterioro en el largo plazo97,99.
Sistema renina-angiotensina-aldosterona
Uno de mecanismos fisiopatológicos más conocidos que intervienene en el síndrome de insuficiencia cardíaca es el SRAA.
La caída del VM cardíaco secundario al daño miocárdico determina una caída de la perfusión renal, sumada a la sobreactivación del sistema nervioso simpático, determinando un incremento de los niveles de renina plasmática, la activación del sistema (Figuras 7, 9, 10 y 11) y, consecuentemente, incremento de los niveles plasmáticos y urinarios de aldosterona100-103.
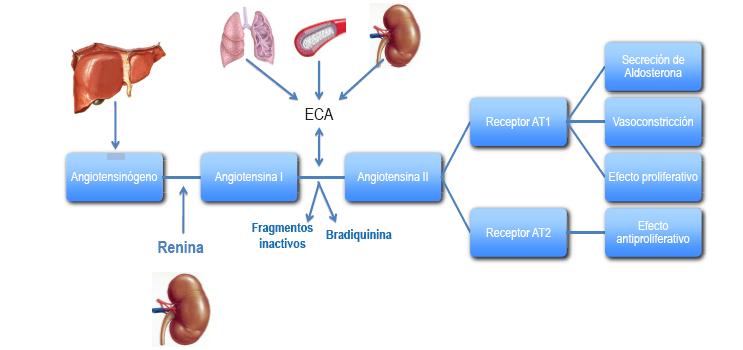
Figura 9. Sistema renina-angiotensina-aldosterona.
ECA: enzima conversora de la angiotensina. AT1: receptor 1 de la angiotensina II. AT2: receptor 2 de la angiotensina II.

Figura 10. Sistema reninaangiotensina. ECA: enzima de conversión de angiotensina. ECA-2: enzima de conversión de angiotensina 2. NEP: neprilisina o endopeptidasa neutra. PEP: prolil endopeptidasa. TOP: thimet oligopeptidasa. APA: aminopeptidasa A. APN: aminopeptidasa N. PreProR: preprorrenina. ProR: pro-renina. AT4: receptor de angiotensina tipo 4. AT1: receptor de angiotensina tipo 1. AT2: receptor de angiotensina tipo 2. Mas: receptor de Ang-(1-7). CarbP: carboxipeptidasa. PO: prolil-oligopeptidasa. CAGE: Chymostatin-sensitive Ang II Generating Enzyme. t-PA: activador tisular del plasminógeno (Tissular Plasminogen Activator).

Figura 11. Sistema renina-angiotensina-aldosterona.
ECA: enzima conversora de la angiotensina. AT1: receptor 1 de la angiotensina II. AT2: receptor 2 de la angiotensina II.
Uno de los avances importantes en el conocimiento de la fisiopatología de la insuficiencia cardíaca se basó en la detección en plasma y orina de una sustancia que poseía la capacidad de retener sodio y agua104-107.
Pronto, se hizo evidente que las etapas iniciales de la insuficiencia cardíaca involucran una expansión del volumen intravascular y aumento de peso, retención de sal y agua, por aumento de la presión hidrostática, generando extravasación de líquido al extravascular (edema)106,108-113 .
Basados en estos hallazgos, los estudios de la hemodinamia renal revelaron una marcada reducción en el flujo sanguíneo renal, determinando una caída en la filtración glomerular, en el esfuerzo por conservar la filtración glomerular o incluso aumentarla102,103,113-116 .
En los pacientes con hiperaldosteronismo, la retención de sodio era evidente no sólo en la orina, sino también en las heces, el sudor y la saliva, lo que indicaba una avidez generalizada de sodio en los pacientes portadores de insuficiencia cardíaca41,108,117-121 .
En la insuficiencia cardíaca, la presencia de mayores cantidades de aldosterona en la orina y en el plasma tienen, al igual que los niveles de catecolaminas, una relación con la supervivencia, mostrando también una estrecha relación con la retención de sodio y agua41,121-125.
Renina
La renina es una enzima que circula en plasma en dos formas moleculares, una de ellas: activa, la renina propiamente dicha, y otra inactiva, de mayor peso molecular: la pro-renina, cuyas proporciones relativas pueden variar de acuerdo con diferentes circunstancias y patologías.
Tanto la renina como su precursor son sintetizados y liberados por las células yuxtaglomerulares en la arteriola aferente renal en respuesta a la disminución del volumen intravascular, con la consecuente caída de la presión de perfusión, detectada por barorreceptores y por una reducción de la concentración de Na+ en la mácula densa.
La producción de renina, también, se ve influenciada por la inervación simpática renal y por varias substancias, entre las que se encuentran la angiotensina II, la vasopresina, las prostaglandinas, los péptidos natriuréticos, el óxido nítrico, la endotelina y la adrenomedulina126-128. También debemos tener en cuenta que, en condiciones normales, los péptidos natriuréticos cardíacos inhiben la secreción de renina.
La renina es la encargada de catalizar la conversión del angiotensinógeno en angiotensina I que, por acción de la enzima conversora de angiotensina (ECA) se convierte en angiotensina II (Figuras 9, 10 y 11).
Angiotensina
Fue descubierta por Eduardo Braun-Menéndez (1903-1959)129 (Figura 12), un notable fisiólogo argentino, nacido en Punta Arenas (Chile), que se naturalizó argentino a temprana edad, criándose en Buenos Aires130. Estudió en la Facultad de Medicina de la Universidad de Buenos Aires (UBA), y eligió medicina cardiovascular y fisiología como sus especialidades. Su tesis doctoral versó sobre la relación entre la glándula pituitaria, el diencéfalo y la presión sanguínea, y fue desarrollada (en 1934) en el Instituto de Fisiología de la Facultad de Medicina de la UBA bajo la supervisión del Premio Nobel en Fisiología (1947), Dr. Bernardo Houssay (Figura 13). Después de recibir su doctorado, fue a Inglaterra para estudiar en The University College London, donde investigó el metabolismo cardíaco. A su regreso de Inglaterra, se unió al prestigioso equipo en el Instituto de Fisiología (UBA), con los Dres. Luis Federico Leloir (futuro Premio Nobel de Bioquímica, 1970), Juan Fasciolo, Juan Muñoz, y Alberto Taquini (Figura 14) para trabajar por algunos años en el mecanismo de hipertensión nefrogénica. Hizo el descubrimiento más importante en su carrera durante esta investigación, que fue la angiotensina, en 1939130. Utilizando la técnica de Goldblatt (pinzamiento de las arterias renales), el equipo formado por Houssay probó que la secreción del riñón (renina) actuaba sobre el plasma sanguíneo produciendo la sustancia (hipertensina, luego llamada angiotensina) que provocaba esa patología. Describe la naturaleza enzimática del sistema renina-angiotensina y su vinculación con la hipertensión. Sobre la base de los experimentos de Goldblatt y colaboradores131 publicados en 1934, Houssay y Fasciolo demostraron un aumento en la presión arterial, colocando el injerto de un riñón isquémico en el cuello de un perro nefrectomizado132. Tras estos resultados iniciales, Houssay y Taquini133 demostraron la presencia de una sustancia presora en la sangre obtenida de la vena renal de un riñón isquémico. Taquini obtuvo una beca en el final de 1938 para trabajar en el Laboratorio de la Fatiga en la Universidad de Harvard con el Dr. B. Dill y el Dr. Paul D. White. Su lugar en el equipo del Instituto de Fisiología (UBA) que estudiaba la isquemia renal fue tomada por el Dr. Eduardo Braun-Menéndez, que acababa de llegar desde el Reino Unido. El grupo argentino, en ese entonces, postuló una reacción de tipo enzima-sustrato134,135; mientras que Page y su grupo, trabajando simultáneamente en Cleveland sobre los mecanismos presores renales, propusieron la existencia en plasma de un activador, como consecuencia de la renina, que intervenía en la producción de una sustancia presora cristalina, la cual llamaron angiotonin136,137. Más tarde, Braun Menéndez, Fasciolo, Leloir, Muñoz y Taquini publican en un libro la obra original del grupo de trabajo del Instituto de Fisiología (UBA)138. Desde entonces, el sistema renina-angiotensina ha sido demostrado estar relacionado con numerosos procesos reguladores fisiológicos, tanto en condiciones normales como fisiopatológicas, desempeñando un papel crítico en la regulación de la circulación, de la HTA, de la insuficiencia cardíaca y de la enfermedad coronaria.

Figura 12. Dr. Eduardo Braun-Menéndez (1903-1959).

Figura 13. Dr. Bernardo Houssay (1887-1971)

Figura 14. Dres. Eduardo Braun Menéndez, Oscar Orías, Bernardo Houssay, Juan Treharme Lewis (abajo de izquierda a derecha) y colaboradores.
La renina plasmática cataliza la hidrólisis del angiotensinógeno (segregado por el hígado) a angiotensina I, el cual es a su vez convertido a angiotensina II (potente vasoconstrictor), por acción de la ECA, la cual se puede encontrar presente en pulmón, endotelio vascular y glomérulos renales, entre otros tejidos (Figura 9).
Actualmente, se conoce que existen otras vías de síntesis de angiotensina II por intermedio de otras enzimas proteolíticas (quimasa, catepsina G), a partir del angiotensinógeno139 (Figura 10).
Los receptores de angiotensina II (AT1 y AT2) poseen diferentes características140-142 (Figura 15), comenzando por el gen para el receptor AT1, el cual está localizado en el cromosoma 3 y, el gen del receptor AT2, localizado en el cromosoma X143-145.
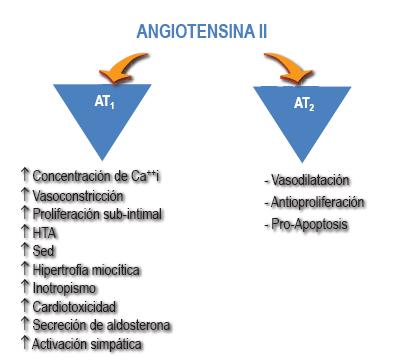
Figura 15. Papel de los receptores de angiotensina. Ca++i: calcio intracelular. HTA: hipertensión arterial.
El AT1 es un receptor de membrana acoplado a la proteína G, localizado en los vasos sanguíneos, riñón, corazón, suprarrenales e hígado.
La unión de la angiotensina II al receptor AT1 determina el acoplamiento a proteínas del subtipo G alfa, desprendiendo la subunidad alfa, que activa la fosfolipasa C, la cual induce incrementos de inositol trifosfato (I3P) y de diacilglicerol (DAG) que activan la proteinquinasa (PKCA) y, en consecuencia, producen un incremento del calcio intracelular (Ca++i)146,147.
En otras células, como la renal o la hepática, el receptor AT1 se acopla a proteínas del subtipo Gi alfa, que actúa inhibiendo la adenilatociclasa y, por lo tanto, reduciendo el AMPc intracelular148-150.
Se han distinguido también dos subtipos de receptores AT1: AT1A y AT1B, que se diferencian en la secuencia de aminoácidos de la zona terminal-C de su estructura molecular151-153.
El receptor AT1A se encuentra expresado principalmente en hígado, riñón, aorta, corazón, pulmón, cerebro, útero, ovario y bazo. Por otra parte, el receptor AT1B se encuentra presente en riñón, hígado, pituitaria, suprarrenales y útero, encontrándose ausente en el corazón, cerebro y bazo141,152,154,155.
La estimulación de los receptores AT1 determina la contracción de la fibra muscular lisa vascular 156 . En el sistema nervioso central, la estimulación de los receptores AT1 promueve la liberación de vasopresina, provocando sed y, en el sistema nervioso periférico, provoca liberación de norepinefrina con la consecuente activación simpática157-159.
A nivel adrenal, la estimulación de los receptores promueve la liberación de catecolaminas y de aldosterona. A nivel renal, contribuye a la mantención de la volemia al promover la vasoconstricción de las arterias preglomerulares, disminución del flujo medular, contracción de las células mesangiales con efecto antidiurético directo a nivel tubular. Además, debemos tener en cuenta que posee un efecto de retroalimentación negativo inhibiendo la renina. A nivel miocárdico, vascular y glomerular, la estimulación de los receptores AT1 posee un efecto hipertrófico y proliferativo, promoviendo la expresión de protooncogenes, factores de crecimiento, la síntesis de ADN y la acumulación de proteínas, mencionándose además un efecto prooxidante y aterogénico.
Por otra parte, los receptores AT2, también, se encuentran en la membrana celular, pero abundan más en el período embrionario (los AT1 son predominantes en la edad adulta), siendo raros en la etapa adulta, se les atribuye una propiedad antiproliferativa que permitiría el balance proliferativo/antiproliferativo con los receptores AT1. Se localizan principalmente en ovario, suprarrenales, cerebro, encontrándose también en las heridas cutáneas o en la neoíntima después de una lesión vascular160,161.
En resumen, podemos decir que la angiotensina II, es uno de los principales efectores del SRAA que posee principalmente dos efectos: vasoconstricción y retención de agua y sodio, consecuencias lógicas del incremento de los niveles de secreción de renina, debidos a fenómenos de hipovolemia y/o hipotensión arterial.
La angiotensina II genera vasoconstricción arteriolar e incremento de las resistencias vasculares que culmina con el aumento de la presión arterial, al tiempo que facilita las acciones del sistema nervioso simpático a diferentes niveles e inhibe el tono vagal. Además de estimular la síntesis y secreción de aldosterona, promueve la reabsorción de Na+ y de agua en el túbulo proximal, aumenta la sed y el apetito por el Na+, dando lugar a diversas respuestas renales directas. Su interacción con el sistema de péptidos natriuréticos es compleja162; así, la infusión de péptido natriurético atrial (PNA) o de péptido natriurético cerebral (PNB) disminuye la producción de renina y de angiotensina II, mientras que la administración de angiotensina II estimula la liberación de PNA y PNB163-166.
Aldosterona
El aumento de los niveles de aldosterona, producido por la estimulación de los receptores de la angiotensina que, en los pacientes con insuficiencia cardíaca se acompaña generalmente de una disminución de la depuración metabólica por un hígado congestivo, determina un incremento de la reabsorción de sodio, contribuyendo además a un remodelamiento vascular y miocárdico (activación de la síntesis de colágeno, hipertrofia miocárdica y vascular)167,168 (Figura 16).

Figura 16. Efectos de la aldosterona. Los mayores niveles de aldosterona provocan un aumento de fi broblastos a nivel cardíaco y vascular, con mayor síntesis de colágeno, más depósito de colágeno en intersticio, más fi brosis miocárdica y vascular, más rigidez ventricular izquierda y vascular, más disfunción ventricular izquierda e incremento de las resistencias vasculares sistémicas (RVS), más insufi ciencia cardíaca y consecuentemente mayor secreción de aldosterona, constituyendo otro de los mecanismos de feed back de la compensación de la insufi ciencia cardíaca, una compensación mal adaptativa, que llega en un momento a provocar más insufi ciencia cardíaca.
La aldosterona, luego de ser sintetizada por la glándula suprarrenal y liberada a la circulación, se une al receptor mineralocorticoideo (RMC) específico, localizado en el citosol de las células epiteliales de los órganos blanco169. La translocación del complejo ligando-receptor al núcleo celular modula la expresión de genes y la transducción de proteínas inducibles que regulan el balance hidroelectrolítico.
La aldosterona posee además acciones no vinculadas directamente a las células epiteliales y no genómicas rápidas, que contribuyen a la homeostasis de la presión arterial. Entre ellas se incluyen, como mencionamos, efectos sobre células endoteliales y tejido cardíaco. Algunas evidencias, además, relacionan a la aldosterona con efectos proinflamatorios en diferentes células no epiteliales, en el contexto de estados de exceso de sodio170-174. Así, se suele dividir a las acciones de la aldosterona en:
- Acciones epiteliales.
- Acciones no epiteliales.
- Mecanismo de acción genómico.
- Mecanismo de acción no genómico.
Acciones epiteliales de la aldosterona
Clásicamente, la aldosterona actúa en las células epiteliales, particularmente en el túbulo colector renal (también posee acción semejante en el colon y en la glándula parótida), regulando el transporte de Na+, K+ y agua.
La acción epitelial de la aldosterona consta de 2 fases, una precoz (1-6 hs) y otra tardía (>6 hs). La fase precoz está mediada por cambios en la expresión génica que activan canales iónicos y proteínas de señal, globalmente denominadas proteínas inducidas por la aldosterona (AIP de sus siglas en ingles: aldosterone induced proteins), estrechamente relacionadas con el transporte electrolítico175.
La fase tardía resulta tanto de efectos primarios como secundarios sobre la expresión génica.
Serum and glucocorticoid-regulated kinase- (Sgk-1)
Dentro de las proteínas inducidas por la aldosterona (AIP), la Sgk-1 (serum and glucocorticoid-regulated kinase-1) es la que ha surgido como el principal intermediario de los efectos de la aldosterona. Se trata de una kinasa de serina-treonina, cuyo mecanismo de acción es aún desconocido. No obstante, ha sido posible establecer que la aldosterona promueve un considerable aumento de la expresión del Sgk-1 en el túbulo renal distal. Es posible que la misma sea responsable de la fosforilación de una proteína reguladora de los canales de sodio epiteliales (ENaC), denominada Nedd4-2-neuronal, induciendo una reducción de su unión al ENaC y dando lugar al aumento de la densidad y de la estabilidad de los canales en la membrana apical, con el consecuente aumento de la reabsorción de Na+ dependiente del ENaC176-181 .
Kirsten Ras GTP-binding protein-2A (Ki-Ras A)
Otra de lasproteínas inducidas por la aldosterona es la expresión de la Ki-Ras A, que tiene lugar durante la fase precoz de su acción. La inducción de la expresión del Ki-Ras A parece necesaria para el efecto de la aldosterona en el transporte de Na+ en las células epiteliales renales. Su sobreexpresión ha sido relacionada con una acción dual aun no explicada, abriendo los canales, pero disminuyendo su cantidad en la membrana plasmática 182-186 .
Fosfatidil inositol 3 kinasa (PI3K)
Esta proteína, una quinasa lipídica, ha sido vinculada con las acciones de la aldosterona, insulina y vasopresina. Si bien no es inducida directamente por la aldosterona, su actividad es aumentada por efecto de ésta y de la insulina. La inhibición de la PI3K reduce tanto las acciones precoces como tardías de la aldosterona, así como también el transporte de Na+ inducido por insulina; además, la estimulación del transporte de Na+ en el riñón por la vasopresina también depende en parte de la PI3K. Por lo tanto, esta quinasa puede representar un punto de convergencia de la activación del ENaC y de la Na+/K+ ATPasa por la aldosterona, la insulina y la vasopresina187-189 .
Factor inducido por hormonas corticoesteroideas (CHIF)
El CHIF es una proteína que se expresa en la membrana de las células epiteliales del colon distal y de los nefrones. Forma parte de una familia de proteínas de transmembrana denominadas FXYD, sigla que representa una secuencia común altamente conservada, y participa de la regulación de los canales iónicos y de las proteínas de transporte, como la subunidad y de la Na+/K+-ATPasa. El mecanismo de transducción de señal es desconocido. Presumiblemente, la aldosterona estimula la expresión del CHIF y la proteína sintetizada interactúa con efectores finales como promotor del transporte190-192 .
Acciones no epiteliales de la aldosterona
El receptor mineralocorticoideo ha sido identificado también en numerosos tejidos no epiteliales, particularmente en el aparato cardiovascular y el sistema nervioso central. No obstante, aunque las propiedades del receptor mineralocorticoideo en esos tejidos son idénticas a las de los correspondientes a tejidos epiteliales, el efecto final es netamente diferente al promover hipertrofia cardíaca, fibrosis y disfunción endotelial vascular y contribuir a la regulación de la tensión arterial, el apetito por la sal y el tono simpático193.
Mecanismo de acción genómico
Como comentáramos, la aldosterona ejerce sus efectos por medio de un receptor citosólico específico, cuyo peso molecular es de 116 Kd, el receptor mineralocorticoideo, que forma parte de la superfamilia de receptores nucleares, está compuesto por varios dominios funcionales entre los que se incluyen118,174,194:
- El dominio N-terminal responsable principalmente de la activación/represión transcripcional.
- El dominio de unión al ADN altamente conservado.
- El dominio C-terminal de unión al ligando que posee la propiedad de reconocimiento de la hormona y de regular los procesos de translocación nuclear.
Este receptor está en el citoplasma celular formando un complejo hétero-oligomérico con las proteínas del choque térmico (HSP, de su nomenclatura inglesa heat-shock protein) y las inmunofilinas. La activación del receptor mineralocorticoideo tras la unión con la aldosterona, genera un cambio conformacional que da lugar a la disociación de las proteínas del complejo, dimerización y translocación al núcleo celular.
El complejo hormona/receptor activado se une a los elementos respondedores a esteroides en el 5 UTR de los genes que responden a la aldosterona activando o reprimiendo la transcripción. Este complejo puede también actuar a través de un mecanismo denominado interferencia o sinergia de la transcripción, al interactuar con otros factores de transcripción que tienen la propiedad de unirse directamente al ADN y activar o suprimir la actividad transcripcional. Este complejo proceso permite una modulación de la transcripción sin necesidad de una interacción directa entre el complejo hormona/receptor y el ADN.
El receptor mineralocorticoideo tiene afinidad similar tanto para los mineralocorticoides como para los glucocorticoides, hecho que se vincula con el alto grado de semejanza estructural entre los receptores de ambos esteroides, a pesar de ello, en condiciones fisiológicas el receptor mineralocorticoideo responde casi exclusivamente a la aldosterona por los efectos de la enzima microsomal, la II b-hidroxiesteroide dehidrogenasa tipo 2 (II b-HSD2), que cataliza la conversión de glucocorticoides activos, capaces de unirse con alta afinidad al receptor mineralocorticoideo, en metabolitos inactivos, es decir la conversión de cortisol a cortisona con escasa afinidad por el receptor mineralocorticoideo, protegiendo así al receptor de la acción de los glucocorticoides circulantes.
Mecanismo de acción no genómico
Los efectos de la aldosterona mediados por su receptor mineralocorticoideo tiene un período de latencia de aproximadamente 45 minutos.
Pero existen efectos rápidos de la aldosterona, también mediados por el receptor mineralocorticoideo, que ocurren dentro de los 15 minutos. Estos efectos tienen lugar en diversos tejidos tanto epiteliales como no epiteliales, tales como los cardiomiocitos, el músculo liso vascular, células endoteliales, renales, epiteliales colónicas y linfocitos. Este efecto no genómico está mediado por diferentes sistemas de segundos mensajeros, en relación con el tipo de célula involucrada173.
Efecto de la aldosterona en el aparato cardiovascular
La expansión del volumen sanguíneo circulante resultante de la retención de agua y sodio, sumados a la acción directa de la aldosterona en el aparato cardiovascular, resultan en un incremento de las cifras de tensión arterial, en respuesta a la hipovolemia e hipotensión arterial41,73.
Por otra parte, la aldosterona induce la proliferación celular (tanto a nivel vascular como cardíaco) y el depósito de colágeno al estimular la síntesis de colágeno tipo I, remodelando el músculo cardíaco y los vasos sanguíneos a expensas de una reducción de la compliance. Estudios recientes muestran que es posible que niveles elevados de aldosterona induzcan el aumento del número de receptores de endotelinas lo que, a su vez, además de favorecer el incremento de la tensión arterial, incrementa también la síntesis de colágeno.
Si bien aun no se encuentra claro el modo por el cual se vinculan la ingesta desmedida de sodio a la exacerbación de los efectos nocivos cardiovasculares de la aldosterona, las acciones nocivas de la aldosterona, también, están estrechamente relacionadas a la ingesta de sodio41. El remodelamiento vascular y cardíaco promovido por la aldosterona está altamente vinculado al sodio, posiblemente debido a una respuesta de activación de bombas de sodio en la membrana celular.
Por otro lado, el crecimiento de los fibroblastos y la síntesis de colágeno involucran a la regulación transcripcional, dependiente de aldosterona, de la NA+/K+ ATPasa, lo cual implica un mecanismo genómico de adaptación lenta, posiblemente, vinculado a factores genéticos de la hipertensión arterial183.
Las características histológicas de la fibrosis cardíaca inducida por la aldosterona incluyen proliferación de los cardiomiocitos y fibroblastos acompañados de un fenómeno de inflamación perivascular intensa. Muchos de los efectos patológicos de la aldosterona requieren de un tiempo prolongado de algunas semanas, por lo que parecen depender del receptor mineralocorticoideo, no obstante ciertos marcadores inflamatorios, como el factor de necrosis tumoral A y el aumento de los depósitos de colágeno tipo III, han sido detectados precozmente, dentro de los dos primeros días, en modelos de fibrosis inducida por mineralocorticoides195,196.
Los receptores mineralocorticoideos han sido identificados en células endoteliales, músculo liso vascular, endocardio y cardiomiocitos, lo cual nos permite adjudicar el remodelamiento cardíaco y vascular a los niveles de aldosterona, más que a la sobrecarga de presión producida por el incremento de la presión arterial.
Niveles elevados de aldosterona se han correlacionado con una serie de efectos adversos sobre el sistema cardiovascular y urinario relacionados con la fibrosis cardíaca y vascular que incluyen: disfunción endotelial, alteraciones de la estructura de la micro y macro vasculatura, disección aórtica, hipertrofia ventricular, deterioro de la función ventricular, tanto sistólica como diastólica, alteraciones de la pared ventricular, prolongación del intervalo PR, microalbuminuria.
La aldosterona se correlaciona no sólo con procesos fibróticos sistémicos, sino también con el incremento de citocinas proinflamatorias como la osteopontina (OPN), llevando el desarrollo fisiopatológico de la insuficiencia cardíaca a un modelo inflamatorio de la misma.
Conclusión
El SRAA es uno de los principales reguladores de la presión sanguínea y del equilibrio hidroelectrolítico -fundamental en el mecanismo de compensación, desarrollo y perpetuación de la insuficiencia cardíaca-, siendo la angiotensina II y la aldosterona los principales mediadores en una diversidad de órganos (corazón, pulmón, cerebro) de funcionamiento endocrino y paracrino.
Estudios recientes aportan evidencias acerca del papel fundamental del SRAA en la función y en la arquitectura cardiovascular, así como en el proceso inflamatorio y en la aterogénesis y, en fin, en una amplia variedad de otros procesos tisulares.
Ha sido demostrado que el SRAA no tiene sólo una función sistémica bajo el control de la renina de origen renal, sino que coexiste con sistemas locales (completos o parciales) activos en una amplia variedad de tejidos. Dichos sistemas podrían no ser completamente independientes unos de otros, sino más bien interactuar entre ellos.
1. Delius L. Cardiovascular regulation disorders and cardiac insufficiency; definition, differential diagnosis and analysis. Medizinische 1957;25:1642-7. [ Links ]
2. Pabst K. Concept and definition of heart failure. Munchener medizinische Wochenschrift 1971;113:1701-8. [ Links ]
3. Wagner S, Cohn K. Heart failure. A proposed definition and classification. Archives of internal medicine 1977;137:675-8. [ Links ]
4. Yasuda H. Cardiac insufficiency: its definition and the mechanism of development. Kango gijutsu : Nursing technique 1978;24:9-17. [ Links ]
5. Gourgon R, Merillon JP, Guiomard A, et al. Left ventricular insufficiency: definition, etiology, physiopathological outline. La Revue du praticien 1982;32:499-507. [ Links ]
6. Denolin H, Kuhn H, Krayenbuehl HP, Loogen F, Reale A. The definition of heart failure. European heart journal 1983;4:445-8. [ Links ]
7. Gourgon R, Cohen Solal A, Himbert D, Aumont MC. Left ventricular insufficiency: definitions and mechanisms. Revue des maladies respiratoires 1987;4:151-8. [ Links ]
8. Ambrosio GB, Riva LM, Zanchi P. Heart failure: problems of definition and clinical staging. Cardiologia 1990;35:707-12. [ Links ]
9. Urbaszek W. Definition and classification of heart failure. Zeitschrift fur Kardiologie 1991;80 Suppl 8:7-12. [ Links ]
10. Szymanski P, Klisiewicz A, Lubiszewska B, et al. Application of classic heart failure definitions of asymptomatic and symptomatic ventricular dysfunction and heart failure symptoms with preserved ejection fraction to patients with systemic right ventricles. The American journal of cardiology 2009;104:414-8. [ Links ]
11. Zannad F, Stough WG, Pitt B, et al. Heart failure as an endpoint in heart failure and non-heart failure cardiovascular clinical trials: the need for a consensus definition. European heart journal 2008;29:413-21. [ Links ]
12. Filippatos G, Zannad F. An introduction to acute heart failure syndromes: definition and classification. Heart failure reviews 2007;12:87-90. [ Links ]
13. Delius L. Cardiovascular regulation disorders and cardiac insufficiency; definition, differential diagnosis and analysis. Die Medizinische 1957;25:1642-7. [ Links ]
14. Marantz PR, Tobin JN, Wassertheil-Smoller S, et al. The relationship between left ventricular systolic function and congestive heart failure diagnosed by clinical criteria. Circulation 1988;77:607-12. [ Links ]
15. Himbert D, Jaeger P, Steg PG, Makowski S, Gourgon R. Cardiac insufficiency. Definition, mechanisms, principles of treatment. Archives des maladies du coeur et des vaisseaux 1990;83:1913-8. [ Links ]
16. Rudolph W. Pathophysiologic and diagnostic aspects of heart failure. Herz 1990;15:147-57. [ Links ]
17. Seward JB, Tajik AJ. Primary cardiomyopathies: classification, pathophysiology, clinical recognition and management. Cardiovascular clinics 1980;10:199-230. [ Links ]
18. Burkart F, Kiowski W. Circulatory abnormalities and compensatory mechanisms in heart failure. The American journal of medicine 1991;90:19S-22S. [ Links ]
19. Heo S, Lennie TA, Okoli C, Moser DK. Quality of life in patients with heart failure: ask the patients. Heart & lung : the journal of critical care 2009;38:100-8. [ Links ]
20. Harding R, Beynon T, Hodson F, et al. Provision of palliative care for chronic heart failure inpatients: how much do we need?. BMC palliative care 2009;8:8. [ Links ]
21. Kiowski W. Compensatory and adaptive mechanisms in congestive heart failure. Journal of cardiovascular pharmacology 1989;14 (Suppl 1):S3-8. [ Links ]
22. Rouleau JL, Kortas C, Bichet D, de Champlain J. Neurohumoral and hemodynamic changes in congestive heart failure: lack of correlation and evidence of compensatory mechanisms. American heart journal 1988;116:746-57. [ Links ]
23. van der Horst IC, de Boer RA, Hillege HL, Boomsma F, Voors AA, van Veldhuisen DJ. Neurohormonal profile of patients with heart failure and diabetes. Netherlands heart journal : monthly journal of the Netherlands Society of Cardiology and the Netherlands Heart Foundation 2010;18:190-6. [ Links ]
24. Watanabe S, Tamura T, Ono K, et al. Insulin-like growth factor axis (insulin-like growth factor-I/insulin-like growth factor-binding protein-3) as a prognostic predictor of heart failure: association with adiponectin. European journal of heart failure 2010;12:1214-22. [ Links ]
25. Yamashita T, Seino Y, Ogawa A, et al. N-terminal pro-BNP is a novel biomarker for integrated cardio-renal burden and early risk stratification in patients admitted for cardiac emergency. Journal of cardiology 2010;55:377-83. [ Links ]
26. Arendt RM, Gerbes AL, Ritter D, Stangl E, Bach P, Zahringer J. Atrial natriuretic factor in plasma of patients with arterial hypertension, heart failure or cirrhosis of the liver. Journal of hypertension Supplement : official journal of the International Society of Hypertension 1986;4:S131-5. [ Links ]
27. Singh V, Martinezclark P, Pascual M, Shaw ES, O'Neill WW. Cardiac biomarkers - the old and the new: a review. Coronary artery disease 2010;21:244-56. [ Links ]
28. Rocchiccioli JP, McMurray JJ, Dominiczak AF. Biomarkers in heart failure: a clinical review. Heart failure reviews 2010;15:251-73. [ Links ]
29. Tsutamoto T, Kawahara C, Nishiyama K, et al. Prognostic role of highly sensitive cardiac troponin I in patients with systolic heart failure. American heart journal 2010;159:63-7. [ Links ]
30. Zairis MN, Tsiaousis GZ, Georgilas AT, et al. Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. International journal of cardiology 2010;141:284-90. [ Links ]
31. de Couto G, Ouzounian M, Liu PP. Early detection of myocardial dysfunction and heart failure. Nature reviews Cardiology 2010;7:334-44. [ Links ]
32. Wang Y, Moreira Mda C, Heringer-Walther S, et al. Plasma ACE2 activity is an independent prognostic marker in Chagas' disease and equally potent as BNP. Journal of cardiac failure 2010;16:157-63. [ Links ]
33. van Kimmenade RR, Mohammed AA, Uthamalingam S, van der Meer P, Felker GM, Januzzi JL, Jr. Red blood cell distribution width and 1-year mortality in acute heart failure. European journal of heart failure 2010;12:129-36. [ Links ]
34. Just H, Drexler H, Hasenfuss G. Pathophysiology and treatment of congestive heart failure. Cardiology 1994;84 (Suppl 2):99-107. [ Links ]
35. Boo JF. Understanding heart failure. Archivos de cardiologia de México 2006;76:431-47. [ Links ]
36. Weissler AM, Harris WS, Schoenfeld CD. Bedside technics for the evaluation of ventricular function in man. The American journal of cardiology 1969;23:577-83. [ Links ]
37. Salcedo EE, Pichard A, Siegel W. Evaluation of left ventricular function by cardiac catheterization, echocardiography, and systolic time intervals. Cleveland Clinic quarterly 1976;43:151-62. [ Links ]
38. Zelis R, Flaim SF, Liedtke AJ, Nellis SH. Cardiocirculatory dynamics in the normal and failing heart. Annual review of physiology 1981;43:455-76. [ Links ]
39. Jacob R, Kissling G. Ventricular pressure-volume relations as the primary basis for evaluation of cardiac mechanics. Return to Frank's diagram. Basic research in cardiology 1989;84:227-46. [ Links ]
40. Yazaki Y. Assessment and evaluation of cardiac function. Nippon Rinsho 1993;51:1177-83. [ Links ]
41. Chaney E, Shaw A. Pathophysiology of fluid retention in heart failure. Contributions to nephrology 2010;164:46-53. [ Links ]
42. Sica DA. Sodium and water retention in heart failure and diuretic therapy: basic mechanisms. Cleveland Clinic journal of medicine 2006;73 (Suppl 2):S2-7; discussion S30-3. [ Links ]
43. Marney AM, Brown NJ. Aldosterone and end-organ damage. Clinical science 2007;113:267-78. [ Links ]
44. Pool PE. The clinical significance of neurohormonal activation. Clinical therapeutics 1997;19 (Suppl A):53-73. [ Links ]
45. Ceconi C, Cargnoni A, Curello S, Ferrari R. Recognized molecular mechanisms of heart failure: approaches to treatment. Portuguese journal of cardiology 1998;17 (Suppl 2):II79-91. [ Links ]
46. Redfield MM, Aarhus LL, Wright RS, Burnett JC, Jr. Cardiorenal and neurohumoral function in a canine model of early left ventricular dysfunction. Circulation 1993;87:2016-22. [ Links ]
47. Francis GS. Neurohumoral mechanisms involved in congestive heart failure. The American journal of cardiology 1985;55:15A-21A. [ Links ]
48. Hannedouche T, Fournier JF. Neurohormonal factors in congestive heart failure. Annales de medecine interne 1987;138:123-9. [ Links ]
49. Vatner SF, Pagani M. Cardiovascular adjustments to exercise: hemodynamics and mechanisms. Progress in cardiovascular diseases 1976;19:91-108. [ Links ]
50. Ertl G, Gaudron P, Eilles C, Schorb W, Kochsiek K. Compensatory mechanisms for cardiac dysfunction in myocardial infarction. Basic research in cardiology 1991;86 (Suppl 3):159-65. [ Links ]
51. Swynghedauw B. Biology of myocardial adaptation to mechanical overload. Biomedicine & pharmacotherapy 1982;36:233-5. [ Links ]
52. Gill RM, Jones BD, Corbly AK, et al. Exhaustion of the Frank-Starling mechanism in conscious dogs with heart failure induced by chronic coronary microembolization. Life sciences 2006;79:536-44. [ Links ]
53. Schwinger RH, Bohm M, Koch A, et al. The failing human heart is unable to use the Frank-Starling mechanism. Circulation research 1994;74:959-69. [ Links ]
54. Kirlin PC, Grekin R, Das S, Ballor E, Johnson T, Pitt B. Neurohumoral activation during exercise in congestive heart failure. The American journal of medicine 1986;81:623-9. [ Links ]
55. Ross JJr., Franklin D, Sasayama S. Preload, afterload, and the role of afterload mismatch in the descending limb of cardiac function. European journal of cardiology 1976;4 (Suppl):77-86. [ Links ]
56. Pasternac A, Noble J, Streulens Y, Lesperance J, Bourassa MG. Myocardial perfusion in myocardiopathies: study at rest and during induced tachycardia. Archives des maladies du coeur et des vaisseaux 1978;71:878-86. [ Links ]
57. Mancia G. Neurohumoral activation in congestive heart failure. American heart journal 1990;120:1532-7. [ Links ]
58. Cosin Aguilar J, Cruz Fernandez JM, de Teresa Galvan E, et al. Neurohormonal factors in heart failure. I. Revista española de cardiología 1996;49:239-52. [ Links ]
59. Cosin Aguilar J, Cruz Fernandez JM, de Teresa Galvan E, et al. Neurohormonal factors in heart failure. II. Revista española de cardiología 1996;49:317-27. [ Links ]
60. Cosin Aguilar J, Cruz Fernandez JM, de Teresa Galvan E, et al. Neurohormonal factors in heart failure (and III). Revista española de cardiología 1996;49:405-22. [ Links ]
61. Rayo I, Marin Huerta E. Chronic heart failure (IV). The neurohormonal factors in chronic heart failure. Their clinical importance and therapeutic implications. Revista española de cardiología 1991;44:656-71. [ Links ]
62. Francis GS, Goldsmith SR, Levine TB, Olivari MT, Cohn JN. The neurohumoral axis in congestive heart failure. Annals of internal medicine 1984;101:370-7. [ Links ]
63. Francis GS, Olivari MT, Goldsmith SR, Levine TB, Pierpont G, Cohn JN. The acute response of plasma norepinephrine, renin activity, and arginine vasopressin to short-term nitroprusside and nitroprusside withdrawal in patients with congestive heart failure. American heart journal 1983;106:1315-20. [ Links ]
64. Goldsmith SR, Francis GS, Cowley AW, Jr, Levine TB, Cohn JN. Increased plasma arginine vasopressin levels in patients with congestive heart failure. Journal of the American College of Cardiology 1983;1:1385-90. [ Links ]
65. Sharma JN, Sharma J. Cardiovascular properties of the kallikrein-kinin system. Current medical research and opinion 2002;18:10-7. [ Links ]
66. Gunaruwan P, Maher A, Williams L, et al. Effects of bradykinin on venous capacitance in health and treated chronic heart failure. Clinical science 2009;116:443-50. [ Links ]
67. Cheng CP, Onishi K, Ohte N, Suzuki M, Little WC. Functional effects of endogenous bradykinin in congestive heart failure. Journal of the American College of Cardiology 1998;31:1679-86. [ Links ]
68. Cugno M, Agostoni P, Brunner HR, Gardinali M, Agostoni A, Nussberger J. Plasma bradykinin levels in human chronic congestive heart failure. Clinical science 2000;99:461-6. [ Links ]
69. Guarda E, Corbalan R, Albertini R, et al. Urinary kallikrein excretion in congestive heart failure. The American journal of cardiology 1991;68:685-7. [ Links ]
70. Yui Y, Kawai C. Effects of prostaglandins and kinin-kallikrein system in heart failure. Nippon Rinsho 1983;41:2735-9. [ Links ]
71. Wang W. Cardiac sympathetic afferent stimulation by bradykinin in heart failure: role of NO and prostaglandins. The American journal of physiology 1998;275:H783-8. [ Links ]
72. Rouleau JL, de Champlain J, Klein M, et al. Activation of neurohumoral systems in postinfarction left ventricular dysfunction. Journal of the American College of Cardiology 1993;22:390-8. [ Links ]
73. Schrier RW. Water and sodium retention in edematous disorders: role of vasopressin and aldosterone. The American journal of medicine 2006;119:S47-53. [ Links ]
74. Benedict CR. Neurohumoral aspects of heart failure. Cardiology clinics 1994;12:9-23. [ Links ]
75. Cohn JN, Levine TB, Francis GS, Goldsmith S. Neurohumoral control mechanisms in congestive heart failure. American heart journal 1981;102:509-14. [ Links ]
76. Mann DL, Cooper Gt. Neurohumoral activation in congestive heart failure: a double-edged sword? Clinical cardiology 1989;12:485-90. [ Links ]
77. Pacher R, Globits S, Bergler-Klein J, et al. Clinical and neurohumoral response of patients with severe congestive heart failure treated with two different captopril dosages. European heart journal 1993;14:273-8. [ Links ]
78. Grassi G, Mancia G. Neurohumoral mechanisms in cardiocirculatory decompensation. Annali italiani di medicina interna: organo ufficiale della Societa italiana di medicina interna 1994;9 (Suppl):61S-7S. [ Links ]
79. Elsner D. Changes in neurohumoral systems during the development of congestive heart failure: impact on cardiovascular and renal function. European heart journal 1995;16 (Suppl N):52-8. [ Links ]
80. Mettauer B, Rouleau JL, Bichet D, et al. Sodium and water excretion abnormalities in congestive heart failure. Determinant factors and clinical implications. Annals of internal medicine 1986;105:161-7. [ Links ]
81. Knotek M, Rogachev B, Ohara M, Schrier RW. Mechanisms of renal retention of sodium and water in heart failure, cirrhosis and pregnancy. Lijecnicki vjesnik 2000;122:20-6. [ Links ]
82. Schrier RW, Fassett RG. Pathogenesis of sodium and water retention in cardiac failure. Renal failure 1998;20:773-81. [ Links ]
83. Schrier RW, Martin PY. Recent advances in the understanding of water metabolism in heart failure. Advances in experimental medicine and biology 1998;449:415-26. [ Links ]
84. Cohn JN. Nervous system control mechanisms in heart failure. Acta medica Scandinavica Supplementum 1986;707:15- 20. [ Links ]
85. Francis GS. Extracardiac features of heart failure: catecholamines and hormonal changes. Cardiology 1988;75 (Suppl 1):19-29. [ Links ]
86. Sukhova ZI, Leont'eva GV, Chechulin Iu S. Adrenal catecholamine secretion during activation of the sympatho-adrenal system. Tsitologiia 1978;20:496-501. [ Links ]
87. Madsen BK, Keller N, Christiansen E, Christensen NJ. Prognostic value of plasma catecholamines, plasma renin activity, and plasma atrial natriuretic peptide at rest and during exercise in congestive heart failure: comparison with clinical evaluation, ejection fraction, and exercise capacity. Journal of cardiac failure 1995;1:207-16. [ Links ]
88. Cohen ML, Schenck KW. Selective down regulation of vascular beta 1 adrenergic receptors after prolonged isoproterenol infusion. Journal of cardiovascular pharmacology 1987;10:365-8. [ Links ]
89. Tse J, Huang MW, Leone RJ, Weiss HR, He YQ, Scholz PM. Down regulation of myocardial beta1-adrenoceptor signal transduction system in pacing-induced failure in dogs with aortic stenosis-induced left ventricular hypertrophy. Molecular and cellular biochemistry 2000;205:67-73. [ Links ]
90. Ceconi C, Curello S, Ferrari R. Catecholamines: the cardiovascular and neuroendocrine system. European heart journal 1998;19 (Suppl F):F2-6. [ Links ]
91. Cleland JG, Dargie HJ. Arrhythmias, catecholamines and electrolytes. The American journal of cardiology 1988;62:55A-9A. [ Links ]
92. Kinugawa T, Ogino K, Osaki S, et al. Prognostic significance of exercise plasma noradrenaline levels for cardiac death in patients with mild heart failure. Circulation journal : official journal of the Japanese Circulation Society 2002;66:261-6. [ Links ]
93. Geslin P, Le Bouil A, Furber A, et al. Plasma noradrenaline and the prognosis of chronic cardiac failure: a multicenter study. Archives des maladies du coeur et des vaisseaux 1998;91:191-9. [ Links ]
94. Watson AM, Hood SG, May CN. Mechanisms of sympathetic activation in heart failure. Clinical and experimental pharmacology & physiology 2006;33:1269-74. [ Links ]
95. Vassalle M, Lin CI. Calcium overload and cardiac function. Journal of biomedical science 2004;11:542-65. [ Links ]
96. Tan LB, Burniston JG, Clark WA, Ng Y, Goldspink DF. Characterization of adrenoceptor involvement in skeletal and cardiac myotoxicity Induced by sympathomimetic agents: toward a new bioassay for beta-blockers. Journal of cardiovascular pharmacology 2003;41:518-25. [ Links ]
97. Brouri F, Findji L, Mediani O, et al. Toxic cardiac effects of catecholamines: role of beta-adrenoceptor downregulation. European journal of pharmacology 2002;456:69-75. [ Links ]
98. Aragona M, Aragona F. Pheochromocytoma and catecholamine cardiomyopathy. Pathologica 1992;84:197-203. [ Links ]
99. Mann DL. Basic mechanisms of disease progression in the failing heart: the role of excessive adrenergic drive. Progress in cardiovascular diseases 1998;41:1-8. [ Links ]
100. Yoshimura M. Aldosterone and heart failure. Nippon Rinsho 2005;63 (Suppl 3):317-22. [ Links ]
101. Yoshimura M. Aldosterone in heart failure. Nippon Yakurigaku Zasshi 2005;126:377-80. [ Links ]
102. Harrison-Bernard LM. The renal renin-angiotensin system. Advances in physiology education 2009;33:270-4. [ Links ]
103. Rea ME, Dunlap ME. Renal hemodynamics in heart failure: implications for treatment. Current opinion in nephrology and hypertension 2008;17:87-92. [ Links ]
104. Tait JF, Simpson SA, Grundy HM. The effect of adrenal extract on mineral metabolism. Lancet 1952;1:122-4. [ Links ]
105. Tait SA, Tait JF, Coghlan JP. The discovery, isolation and identification of aldosterone: reflections on emerging regulation and function. Mol Cell Endocrinol 2004;217:1-21. [ Links ]
106. Zannad F. Aldosterone and heart failure. European heart journal 1995;16 (Suppl N):98-102. [ Links ]
107. Weber KT, Villarreal D. Role of aldosterone in congestive heart failure. Postgraduate medicine 1993;93:203-7, 211-212, 216-218 passim. [ Links ]
108. Papper S. Sodium and water: an overview. The American journal of the medical sciences 1976;272:43-51. [ Links ]
109. Klahr S, Slatopolsky E. Renal regulation of sodium excretion. Function in health and in edema-forming states. Archives of internal medicine 1973;131:780-91. [ Links ]
110. Weber KT, Villarreal D. Aldosterone and antialdosterone therapy in congestive heart failure. The American journal of cardiology 1993;71:3A-11A. [ Links ]
111. Selektor Y, Weber KT. The salt-avid state of congestive heart failure revisited. The American journal of the medical sciences 2008;335:209-18. [ Links ]
112. Marenzi GC, Grazi S, Salvioni A, et al. The factors that regulate water-salt metabolism in congestive heart failure. Cardiología 1993;38:287-95. [ Links ]
113. Makaritsis KP, Liakopoulos V, Leivaditis K, Eleftheriadis T, Stefanidis I. Adaptation of renal function in heart failure. Renal failure 2006;28:527-35. [ Links ]
114. Riley DJ, Weir M, Bakris GL. Renal adaptation to the failing heart. Understanding the cascade of responses. Postgraduate medicine 1994;95:141-6, 9-50. [ Links ]
115. Ritz E, Fliser D. The kidney in congestive heart failure. European heart journal 1991;12 (Suppl C):14-20. [ Links ]
116. Hollenberg NK. Control of renal perfusion and function in congestive heart failure. The American journal of cardiology 1988;62:72E-5E. [ Links ]
117. Wilson DR, Laidlaw JC, Ruse JL. Fecal and salivary electrolytes in the diagnosis of primary aldosteronism. Can Med Assoc J 1971;105:1300-5. [ Links ]
118. Ronconi V, Giacchetti G, Boscaro M. Endocrinology of aldosterone. Italian heart journal: official journal of the Italian Federation of Cardiology 2005;6 (Suppl 1):5S-15S. [ Links ]
119. Smilari L, Licitra G, Zappia A. Pathogenetic problems in congestive cardiac insufficiency. IV. The electrolytes (Na+, K+) in sweat induced by heat and in urine of normal subjects exposed to various stimuli (aldosterone, sodium chloride). Archivio per le scienze mediche 1966;121:328-42. [ Links ]
120. Bansal S, Lindenfeld J, Schrier RW. Sodium retention in heart failure and cirrhosis: potential role of natriuretic doses of mineralocorticoid antagonist? Circulation Heart failure 2009;2:370-6. [ Links ]
121. Hensen J, Abraham WT, Durr JA, Schrier RW. Aldosterone in congestive heart failure: analysis of determinants and role in sodium retention. American journal of nephrology 1991;11:441-6. [ Links ]
122. Marenzi G, Lauri G, Guazzi M, et al. Cardiac and renal dysfunction in chronic heart failure: relation to neurohumoral activation and prognosis. The American journal of the medical sciences 2001;321:359-66. [ Links ]
123. Eiskjaer H, Bagger JP, Danielsen H, et al. Mechanisms of sodium retention in heart failure: relation to the reninangiotensin-aldosterone system. The American journal of physiology 1991;260:F883-9. [ Links ]
124. Francis GS. Neuroendocrine manifestations of congestive heart failure. The American journal of cardiology 1988;62:9A-13A. [ Links ]
125. Hillege HL, Girbes AR, de Kam PJ, et al. Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation 2000;102:203-10. [ Links ]
126. Meune C, Mourad JJ, Bergmann JF, Spaulding C. Interaction between cyclooxygenase and the renin-angiotensinaldosterone system: rationale and clinical relevance. JRAAS 2003;4:149-54. [ Links ]
127. Schrier RW, Abraham WT. Hormones and hemodynamics in heart failure. The New England journal of medicine 1999;341:577-85. [ Links ]
128. Yang RF, Yin JX, Li YL, Zimmerman MC, Schultz HD. Angiotensin-(1-7) Increases Neuronal Potassium Current via a Nitric Oxide-Dependent Mechanism. Am J Physiol Cell Physiol 2010. [ Links ]
129. Jaim Etcheverry G. La concepción universitaria de Eduardo Braun Menéndez. MEDICINA (Buenos Aires) 2000; 60: 149-154. [ Links ]
130. A In Memoriam. Hypertension 1998; 32: 1-2. [ Links ]
131. Goldblatt H, Lynch J, Hanzal RF, Summerville WW. Studies on experimental hypertension: the production of persistent elevation of systolic blood pressure by means of renal ischemia. J Exp Med 1934;59:347. [ Links ]
132. Houssay BA, Fasciolo JC. Secreción hipertensora del riñón isquemiado. Rev Soc Argent Biol 1937;13:284. [ Links ]
133. Houssay BA, Taquini AC. Acción vasoconstrictora de la sangre venosa del riñón isquemiado. Rev Soc Argent Biol 1938;14:5. [ Links ]
134. Braun-Menéndez E, Fasciolo JC, Leloir LF, Muñoz JM. La substancia hipertensora de la sangre del riñón isquemiado. Rev Soc Argent Biol 1939;15:420. [ Links ]
135. Braun-Menéndez E, Fasciolo JC, Leloir LF, Muñoz JM. The substance causing renal hypertension. J Physiol 1940;98:283. [ Links ]
136. Page IH, Helmer OM. A crystalline pressor substance, angiotonin, resulting from the reaction between renin and renin activator. Proc Soc Clin Invest 1939;12:17. [ Links ]
137. Page IH, Helmer OM. A crystalline pressor substance, angiotonin, resulting from the reaction between renin and renin activator. J Exp Med 1940;71:29. [ Links ]
138. Braun-Menéndez E, Fasciolo JC, Leloir LF, Muñoz JM, Taquini AC. Hipertensión Arterial Nefrógena. Buenos Aires, Argentina: El Ateneo; 1943. [ Links ]
139. Husain A. The chymase-angiotensin system in humans. J Hypertens 1993;11:1155-9. [ Links ]
140. Wolf G, Neilson EG. From converting enzyme inhibition to angiotensin II receptor blockade: new insight on angiotensin II receptor subtypes in the kidney. Exp Nephrol 1996;4 Suppl 1:8-19. [ Links ]
141. Edwards RM, Aiyar N. Angiotensin II receptor subtypes in the kidney. J Am Soc Nephrol 1993;3:1643-52. [ Links ]
142. Sechi LA, Grady EF, Griffin CA, Kalinyak JE, Schambelan M. Distribution of angiotensin II receptor subtypes in rat and human kidney. Am J Physiol 1992;262:F236-40. [ Links ]
143. Szpirer C, Riviere M, Szpirer J, et al. Chromosomal assignment of human and rat hypertension candidate genes: type 1 angiotensin II receptor genes and the SA gene. J Hypertens 1993;11:919-25. [ Links ]
144. Koike G, Horiuchi M, Yamada T, Szpirer C, Jacob HJ, Dzau VJ. Human type 2 angiotensin II receptor gene: cloned, mapped to the X chromosome, and its mRNA is expressed in the human lung. Biochem Biophys Res Commun 1994;203:1842-50. [ Links ]
145. Lazard D, Briend-Sutren MM, Villageois P, Mattei MG, Strosberg AD, Nahmias C. Molecular characterization and chromosome localization of a human angiotensin II AT2 receptor gene highly expressed in fetal tissues. Receptors Channels 1994;2:271-80. [ Links ]
146. Lassegue B, Alexander RW, Clark M, Griendling KK. Angiotensin II-induced phosphatidylcholine hydrolysis in cultured vascular smooth-muscle cells. Regulation and localization. Biochem J 1991;276 (Pt 1):19-25. [ Links ]
147. Suzuki A, Shinoda J, Oiso Y, Kozawa O. Tyrosine kinase is involved in angiotensin II-stimulated phospholipase D activation in aortic smooth muscle cells: function of Ca2+ influx. Atherosclerosis 1996;121:119-27. [ Links ]
148. Klett C, Nobiling R, Gierschik P, Hackenthal E. Angiotensin II stimulates the synthesis of angiotensinogen in hepatocytes by inhibiting adenylylcyclase activity and stabilizing angiotensinogen mRNA. J Biol Chem 1993;268:25095-107. [ Links ]
149. Douglas JG. Angiotensin receptor subtypes of the kidney cortex. Am J Physiol 1987;253:F1-7. [ Links ]
150. Madhun ZT, Ernsberger P, Ke FC, Zhou J, Hopfer U, Douglas JG. Signal transduction mediated by angiotensin II receptor subtypes expressed in rat renal mesangial cells. Regul Pept 1993;44:149-57. [ Links ]
151. Chiu AT, McCall DE, Ardecky RJ, Duncia JV, Nguyen TT, Timmermans PB. Angiotensin II receptor subtypes and their selective nonpeptide ligands. Receptor 1990;1:33-40. [ Links ]
152. Wong PC, Chiu AT, Duncia JV, Herblin WF, Smith RD, Timmermans PB. Angiotensin II receptor antagonists and receptor subtypes. Trends in endocrinology and metabolism: TEM 1992;3:211-7. [ Links ]
153. Timmermans PB, Chiu AT, Herblin WF, Wong PC, Smith RD. Angiotensin II receptor subtypes. Am J Hypertens 1992;5:406-10. [ Links ]
154. Kakar SS, Sellers JC, Devor DC, Musgrove LC, Neill JD. Angiotensin II type-1 receptor subtype cDNAs: differential tissue expression and hormonal regulation. Biochem Biophys Res Commun 1992;183:1090-6. [ Links ]
155. de Gasparo M, Rogg H, Brink M, et al. Angiotensin II receptor subtypes and cardiac function. European heart journal 1994;15 (Suppl D):98-103. [ Links ]
156. Timmermans PB, Smith RD. Angiotensin II receptor subtypes: selective antagonists and functional correlates. European heart journal 1994;15 (Suppl D):79-87. [ Links ]
157. Rowe KD, Schwartz JA, Lomax LL, Knuepfer MM. Central angiotensin II receptors mediate hemodynamic response variability to stressors. Am J Physiol Regul Integr Comp Physiol 2006;291:R719-27. [ Links ]
158. Watanabe T, Barker TA, Berk BC. Angiotensin II and the endothelium: diverse signals and effects. Hypertension 2005;45:163-9. [ Links ]
159. Saavedra JM. Brain angiotensin II: new developments, unanswered questions and therapeutic opportunities. Cell Mol Neurobiol 2005;25:485-512. [ Links ]
160. Barker TA, Massett MP, Korshunov VA, Mohan AM, Kennedy AJ, Berk BC. Angiotensin II type 2 receptor expression after vascular injury: differing effects of angiotensin-converting enzyme inhibition and angiotensin receptor blockade. Hypertension 2006;48:942-9. [ Links ]
161. Eto H, Biro S, Miyata M, et al. Angiotensin II type 1 receptor participates in extracellular matrix production in the late stage of remodeling after vascular injury. Cardiovasc Res 2003;59:200-11. [ Links ]
162. Fitzpatrick MA, Nicholls MG, Espiner EA, Ikram H, Bagshaw P, Yandle TG. Neurohumoral changes during onset and offset of ovine heart failure: role of ANP. The American journal of physiology 1989;256:H1052-9. [ Links ]
163. Dogan A, Gedikli O, Ozaydin M, Acar G. Plasma renin activity and pro-B-type natriuretic peptide levels in different atrial fibrillation types. Anadolu Kardiyol Derg 2010;10:317-22. [ Links ]
164. Chen HH, Cataliotti A, Schirger JA, Martin FL, Harstad LK, Burnett JC, Jr. Local renal delivery of a natriuretic peptide a renal-enhancing strategy for B-type natriuretic peptide in overt experimental heart failure. Journal of the American College of Cardiology 2009;53:1302-8. [ Links ]
165. Burger AJ. A review of the renal and neurohormonal effects of B-type natriuretic peptide. Congestive heart failure 2005;11:30-8. [ Links ]
166. Suttner SW, Boldt J. Natriuretic peptide system: physiology and clinical utility. Current opinion in critical care 2004;10:336-41. [ Links ]
167. Crabbe J, Thorn GW. Effects of Aldosterone on the Renal Function of the Normal Human. Rev Fr Etud Clin Biol 1964;9:729-35. [ Links ]
168. Smilari L, Licitra G, Zappia A. Pathogenetic problems of congestive failure. II. Effects of aldosterone on electrolytes (Na+ and K+) in the saliva and urine of normal subjects and decompensated heart patients. Archivio per le scienze mediche 1966;121:35-50. [ Links ]
169. Rocha R, Stier CT, Jr. Pathophysiological effects of aldosterone in cardiovascular tissues. Trends in endocrinology and metabolism: TEM 2001;12:308-14. [ Links ]
170. Harvey BJ, Higgins M. Nongenomic effects of aldosterone on Ca2+ in M-1 cortical collecting duct cells. Kidney Int 2000;57:1395-403. [ Links ]
171. Losel RM, Feuring M, Falkenstein E, Wehling M. Nongenomic effects of aldosterone: cellular aspects and clinical implications. Steroids 2002;67:493-8. [ Links ]
172. Gunaruwan P, Struthers A, Frenneaux M. Looking at nongenomic vascular effects of aldosterone. Atherosclerosis 2004;173:135. [ Links ]
173. Chai W, Garrelds IM, de Vries R, Batenburg WW, van Kats JP, Danser AH. Nongenomic effects of aldosterone in the human heart: interaction with angiotensin II. Hypertension 2005;46:701-6. [ Links ]
174. Yoshida T, Shin-ya H, Nakai S, et al. Genomic and nongenomic effects of aldosterone on the individual variation of the sweat Na+ concentration during exercise in trained athletes. Eur J Appl Physiol 2006;98:466-71. [ Links ]
175. Blazer-Yost B, Geheb M, Preston A, Handler J, Cox M. Aldosterone-induced proteins in renal epithelia. Biochim Biophys Acta 1982;719:158-61. [ Links ]
176. Verrey F, Summa V, Heitzmann D, et al. Short-term aldosterone action on Na,K-ATPase surface expression: role of aldosterone- induced SGK1? Ann N Y Acad Sci 2003;986:554-61. [ Links ]
177. Rozansky DJ, Cornwall T, Subramanya AR, et al. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 2009;119:2601-12. [ Links ]
178. Terada Y, Kuwana H, Kobayashi T, et al. Aldosteronestimulated SGK1 activity mediates profibrotic signaling in the mesangium. J Am Soc Nephrol 2008;19:298-309. [ Links ]
179. Lee IH, Campbell CR, Cook DI, Dinudom A. Regulation of epithelial Na+ channels by aldosterone: role of Sgk1. Clin Exp Pharmacol Physiol 2008;35:235-41. [ Links ]
180. Ring AM, Leng Q, Rinehart J, et al. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci U S A 2007;104:4025-9. [ Links ]
181. McCormick JA, Bhalla V, Pao AC, Pearce D. SGK1: a rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology (Bethesda) 2005;20:134-9. [ Links ]
182. Williams R. The year's successes in failure: Circulation Research takes a look at the key research developments of 2009 that are providing hope in the field of heart failure.Circulation research 2010;106:213-5. [ Links ]
183. Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol 2003;284:H176-84. [ Links ]
184. Huang S, Zhang A, Ding G, Chen R. Aldosterone-induced mesangial cell proliferation is mediated by EGF receptor transactivation. Am J Physiol Renal Physiol 2009;296:F1323-33. [ Links ]
185. Terada Y, Kobayashi T, Kuwana H, et al. Aldosterone stimulates proliferation of mesangial cells by activating mitogenactivated protein kinase 1/2, cyclin D1, and cyclin A. J Am Soc Nephrol 2005;16:2296-305. [ Links ]
186. Hendron E, Stockand JD. Activation of mitogen-activated protein kinase (mitogen-activated protein kinase/extracellular signal-regulated kinase) cascade by aldosterone. Mol Biol Cell 2002;13:3042-54. [ Links ]
187. Helms MN, Liu L, Liang YY, et al. Phosphatidylinositol 3,4,5-trisphosphate mediates aldosterone stimulation of epithelial sodium channel (ENaC) and interacts with gamma- ENaC. J Biol Chem 2005;280:40885-91. [ Links ]
188. Tsybouleva N, Zhang L, Chen S, et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 2004;109:1284-91. [ Links ]
189. Musch MW, Lucioni A, Chang EB. Aldosterone regulation of intestinal Na absorption involves SGK-mediated changes in NHE3 and Na+ pump activity. Am J Physiol Gastrointest Liver Physiol 2008;295:G909-19. [ Links ]
190. Wong S, Brennan FE, Young MJ, Fuller PJ, Cole TJ. A direct effect of aldosterone on endothelin-1 gene expression in vivo. Endocrinology 2007;148:1511-7. [ Links ]
191. Wald H, Popovtzer MM, Garty H. Differential regulation of CHIF mRNA by potassium intake and aldosterone. Am J Physiol 1997;272:F617-23. [ Links ]
192. Wald H, Goldstein O, Asher C, Yagil Y, Garty H. Aldosterone induction and epithelial distribution of CHIF. Am J Physiol 1996;271:F322-9. [ Links ]
193. Funder JW. Aldosterone and mineralocorticoid receptors in the cardiovascular system. Progress in cardiovascular diseases 2010;52:393-400. [ Links ]
194. Gunaruwan P, Schmitt M, Sharman J, Lee L, Struthers A, Frenneaux M. Effects of aldosterone on forearm vasculature in treated chronic heart failure. The American journal of cardiology 2005;95:412-4. [ Links ]
195. Lijnen P, Petrov V. Induction of cardiac fibrosis by aldosterone. Journal of molecular and cellular cardiology 2000;32:865-79. [ Links ]
196. Brilla CG. Aldosterone and myocardial fibrosis in heart failure. Herz 2000;25:299-306. [ Links ]

