










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkInsuficiencia cardíaca
versión On-line ISSN 1852-3862
Insuf. card. vol.9 no.3 Ciudad Autónoma de Buenos Aires set. 2014
ARTÍCULO DE ACTUALIZACION
Feocromocitoma
Cecilia Perel1
1 Médica cardióloga universitaria. Especialista en Hipertensión Arterial. Miembro de The American Heart Association y The American Stroke Association. Council on High Blood Pressure Research (AHA). Ciudad Autónoma de Buenos Aires. República Argentina.
Correspondencia: Dra. Cecilia Perel.
Soler 4567. CP 1425. Ciudad de Buenos Aires. República Argentina.
Tel/fax: 54-11-4864-8998
E-mail:info@ceciliaperel.com
Recibido: 25/02/2014
Aceptado: 16/05/2014
Resumen
El feocromocitoma (Feo) y el paraganglioma (PGL) son tumores endocrinos de rara aparición que derivan de la médula suprarrenal o de la cresta neuronal, conocidos como paraganglia. El Feo o el PGL pueden surgir en forma esporádica o ser parte de un síndrome tumoral hereditario. Estos tumores se originan por la mutación en los genes VHL, RET, también pueden comprometer genes que se encuentran involucrados como parte de síndromes. La clínica dependerá de la funcionalidad o del predominio simpático del Feo/PGL. Los signos y los síntomas del tumor simpático cromafín incluyen hipertensión arterial sostenida o en episodios paroxísticos. Presenta la clásica triada de cefalea, palpitaciones y diaforesis de aparición paroxística, palidez, hipotensión ortostática y síncope, temblor, ansiedad. Así también como, dolor precordial, arritmias, hipertrofia cardíaca, cardiomiopatía, hasta evolucionar a insuficiencia cardíaca. El diagnóstico del Feo/PGL ha sido simplificado gracias a los avances en los estudios para detectar y cuantificar los niveles de catecolaminas y sus metabolitos, tanto en sangre como en orina. La localización del tumor puede ser a través de tomografía computada y resonancia magnética nuclear, ambos métodos muy útiles. Los test funcionales utilizan la centellografía nuclear con metaiodobenzilguanidina (MIBG), siendo altamente específicos para confirmar la secreción natural de catecolaminas por el tumor. Se realiza tratamiento preparatorio para la resección quirúrgica del tumor, iniciándose 1 a 2 semanas antes de la cirugía. El pronóstico del tumor resecado en su totalidad es muy bueno. Hasta la actualidad no se dispone de un sistema de estadificación para el Feo/PGL maligno y la expectativa de vida dependerá de la ubicación de las metástasis (MTS), con una sobrevida menor a 5 años en los pacientes que presentan MTS a nivel pulmonar o hepático. También puede observarse un síndrome símil a una cardiomiopatía por estrés (Tako-tsubo), es decir que el Feo es capaz de producir una cardiomiopatía reversible. El diagnóstico precoz del Feo/PGL puede reducir su morbi-mortalidad, y si no es diagnosticado puede llevar a la muerte del paciente.
Palabras clave: Feocromocitoma ; Paraganglioma ; Hipertensión arterial ; Médula suprarrenal ; Cromafín ; Catecolaminas
Summary
Pheochromocytoma
Pheochromocytoma (Pheo) and paraganglioma (PGL) are rarely seen endocrine tumors derived from the adrenal medulla or the neuronal crest known as paraganglia. The Pheo or PGL may emerge sporadically or be part of a hereditary tumor syndrome. These tumors originate by mutation in the VHL, RET genes, and may also compromise genes that are involved as part of syndromes. The clinic will depend on the functionality or sympathetic predominance of Pheo/PGL. Signs and symptoms of sympathetic chromaffin tumor include sustained or paroxysmal hypertension episodes. Present the classic triad of headache, palpitations and diaphoresis of paroxysmal onset, pallor, orthostatic hypotension and syncope, tremor, anxiety. So also as chest pain, arrhythmias, cardiac hypertrophy, cardiomyopathy, to evolve at heart failure.
The diagnosis of Pheo/PGL has been simplified by advances in studies to detect and quantify the levels of catecholamines and their metabolites in blood and urine. The location of the tumor can be through computed tomography and magnetic resonance imaging useful methods. The functional test using nuclear scintigraphy with metaiodobenzylguanidine (MIBG) is highly specific to confirm the natural secretion of catecholamines by the tumor. Pre-treatment is performed for surgical resection of the tumor, starting 1 to 2 weeks prior to surgery. The prognosis of the resected tumor as a whole is very good. To date there is no staging system for Pheo/PGL malignant and life expectancy depend on the location of metastases (MTS), with a lower 5-year survival in patients with pulmonary or hepatic MTS level. May also be observed a simile syndrome stress cardiomyopathy (Tako-tsubo), i.e. the Pheo is able to produce a reversible cardiomyopathy. Early diagnosis of Pheo/PGL may reduce your morbidity and mortality if not diagnosed and may lead to death.
Keywords:Pheochromocytoma ; Paraganglioma ; Hypertension ; Adrenal medulla ; Chromaffin ; Catecholamines
Resumo
Feocromocitoma
Feocromocitoma (Feo) e paraganglioma (PGL) são tumores neuroendócrinos de rara aparição derivados da medula supra-renal ou da crista neural conhecido como paragânglia. O Feo ou o PGL pode ocorrer esporadicamente ou ser parte de uma síndrome tumoral hereditária. Estes tumores são causados por mutações nos genes VHL, RET, também pode implicar genes que estão envolvidos, como parte de síndromes. A clínica vai depender da funcionalidade ou a predominância do simpático de Feo/PGL. Sinais e sintomas de tumor cromafin simpático incluem hipertensão sustentada ou episódios paroxísticos. Têm a tríade clássica de dor de cabeça, palpitações e sudorese de início paroxística, palidez, hipotensão ortostática e síncope, tremor, ansiedade. Assim também como dor torácica, arritmias, hipertrofia cardíaca, cardiomiopatia, de progredir para insuficiência cardíaca. O diagnóstico de Feo/PGL foi simplificado pelos avanços nos estudos para detectar e quantificar os níveis de catecolaminas e seus metabólitos no sangue e na urina. A localização do tumor pode ser através de tomografia computadorizada e ressonância magnética, métodos úteis. Os testes funcionais usando cintigrafia de corpo com metaiodobenzilguanidina (MIBG) são altamente específicos para confirmar a secreção natural de catecolaminas pelo tumor. O pré-tratamento é realizado durante a ressecção cirúrgica do tumor, a partir de 1 ou 2 semanas antes da cirurgia. O prognóstico do tumor ressecado como um todo é muito bom. Até o momento não há nenhum sistema de estadiamento para Feo/PGL maligno e a expectativa de vida vai depender da localização das metástases (MTS), com uma menor sobrevida em 5 anos em pacientes com MTS nos pulmões ou fígado.
Também pode ser observada uma síndrome símile na cardiomiopatia de estresse (Tako-tsubo), ou seja, o Feo é capaz de produzir uma cardiomiopatia reversível. O diagnóstico precoce de Feo/PGL pode reduzir a morbidade e mortalidade, e se não diagnosticada pode levar à morte.
Palavras-chave:Feocromocitoma ; Paraganglioma ; Hipertensão ; Medula adrenal ; Cromafin ; Catecolaminas
Introducción
El feocromocitoma (Feo) y el paraganglioma (PGL) son tumores endocrinos de rara aparición, que derivan de la médula suprarrenal (Figura 1) o de la cresta neuronal conocidos como paraganglia (tejido especializado de la cresta neutral asociado a los nervios del sistema nervioso autónomo, actuando algunos como quimiorreceptores, localizados en la bifurcación carotídea y que pueden detectar cambios en el pH sanguíneo, presión de O2 y CO2, cambios en la presión arterial, ritmo respiratorio o cardíaco; únicos o múltiples se asocian a paragangliomas en otros sitios del cuerpo), pudiendo aparecer en cualquier localización que se encuentre paraganglia. Comprenden menos del 7% de los tumores del sistema nervioso simpático, con una incidencia estimada de 0,3 casos por millón por año1,2. Aproximadamente entre el 10% y el 20% de los casos se diagnostican durante la niñez, con una edad promedio de 11 años, con un leve predominio en los varones.
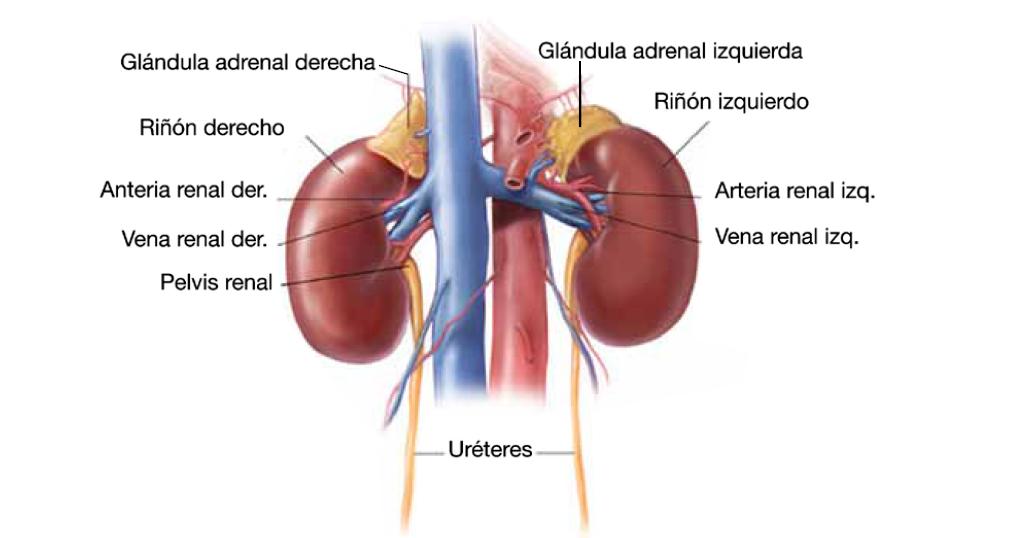
Figura 1. Localización anatómica de las glándulas suprarrenales.
Cuando un niño presenta hipertensión arterial (HTA), en el 1,7%, tiene un tumor que secreta catecolaminas3.
El Feo o PGL puede surgir en forma esporádica o hereditaria.
El PGL puede ser funcionante (simpático) o generalmente no funcionante (parasimpático), eso dependerá del sitio de origen y de los eventos mutacionales subyacentes4-6. La gran mayoría de los PGL que surgen a nivel de la cabeza o del cuello son no funcionantes, mientras que los PGL de origen abdominal son tumores secretantes cromafines (Figura 2). En ciertos casos el nombre de PGL o de Feo pueden ser indistintamente utilizados, tal como es el caso de los ganglioneuroblastomas, ganglioneuromas o del carcinoma neuroendocrino7,8.

Figura 2. El sistema paraganglial y sitios topográficos (en rojo) de los feocromocitomas y paragangliomas. A. Feocromocitoma adrenal. B. Feocromocitoma extra adrenal. C. Paraganglioma de cabeza y cuello.
Los múltiples efectos de las catecolaminas dan el resultado de una amplia variedad de presentaciones clínicas.
El Feo y los PGL funcionantes son tumores productores de catecolaminas, también llamados tumores cromafines. Entre el 35% al 40%, posee una base hereditaria secundaria a una mutación de las líneas germinales 9,10 , por lo que al presente son los tumores más frecuentes hereditarios. Tanto la forma hereditaria como la esporádica difieren marcadamente en cuanto al contenido de catecolaminas, variando el perfil hormonal tanto urinario como plasmático.
El Feo es el término que se utiliza para denominar al tumor secretante de la médula adrenal, mientras que los PGL son tumores extra adrenales que surgen de la paraganglia simpática y parasimpática. Los PGL pueden aparecer desde la base del cráneo hasta la pelvis, pero la gran mayoría se localizan en la cabeza, en el cuello o a nivel abdominal cerca de los vasos renales o en el órgano de Zuckerland, que está localizado alrededor del origen de la arteria mesentérica inferior, y es la colección extra adrenal más grande de tejido cromafín (Figura 3). El término de Feo muchas veces se utiliza indistintamente con el de PGL, pero lo correcto es mantener la separación entre los dos tipos tumorales debido a las diferencias genéticas, a la clínica y al potencial maligno.

Figura 3. Localización de potenciales ubicaciones de feocromocitomas.
Aproximadamente, el 50% de los tumores produce una mezcla de noradrenalina y adrenalina, el resto produce casi exclusivamente noradrenalina, y raramente el tumor produce principalmente dopamina11,12.
Los PGL simpáticos son generalmente no secretores y se localizan en la cabeza y en el cuello (Figura 2C). Hace una década sólo un 10% de los Feo/PGL eran considerados hereditarios.
Debido a que la secreción de catecolaminas puede variar marcadamente, el relativo aumento de las concentraciones de adrenalina y noradrenalina dan un pobre reflejo de los contenidos tumorales de catecolaminas. En cambio, los metabolitos metilados como la normetanefrina y metanefrina son sintetizados continuamente por las células tumorales. Por ende, las concentraciones de estos metabolitos por arriba del valor normal dan una relación altamente positiva del tejido tumoral de adrenalina y noradrenalina.
La enfermedad de Von Hippel Lindau (VHL) se caracteriza por un aumento exclusivo de normetanefrina, reflejando la producción de noradrenalina13. Por el contrario, los pacientes portadores de una neoplasia endócrina múltiple 2 (NEM 2), debida a una mutación del gen RET, desarrollan un tumor que se caracteriza por el aumento de metanefrina plasmática, indicando por ende una producción de adrenalina por parte del tumor. Como puede observarse, estas diferencias se hallan en relación con los distintos perfiles de expresión genética.
La edad de aparición difiere también si es hereditario o esporádico, siendo en el primer caso, la edad promedio de aparición entre 12 y 19 años, antes que los PGL esporádicos.
Aspectos genéticos
Ya mencionado anteriormente, el Feo o PGL puede surgir en forma esporádica o ser parte de un síndrome tumoral hereditario. Estos tumores se originan por la mutación en los genes VHL, RET y a genes que se encuentran como parte de síndromes: la enfermedad de VHL y la NEM 2 A y B, el síndrome familiar de PGL y más raramente de la neurofibromatosis tipo 1, del NEM 1 y de la esclerosis tuberosa11,14-19, o de una reciente y nueva mutación de la línea germinal en el dominio 2 del gen de la prolina hidroxilasa20. Finalmente, la asociación de un PGL no familiar, un tumor gastrointestinal y de un condroma pulmonar (triada de Carney), se ha descripto en un pequeño número de pacientes, pero aún falta determinar la etiología genética21,22. Estos genes codifican a cuatro subunidades de la succinil dehidrogenasa (SDHA, SDHB, SDHC y SDHD), el gen responsable por la flavonización de la SDHA (SDHFA2), y los recientes genes descubiertos: el TMEM127 y el MAX. Las mutaciones en los genes de la SDH pueden producir tanto Feo como PGL.
Dentro de las mutaciones del gen SDH, las más frecuentes son la del SDHB y la del SDHD. Los síndromes se denominan PGL4 y PGL1, respectivamente. El PGL4 se manifiesta principalmente en territorio abdominal, tiene un alto grado de malignidad, alrededor de un 40% de las mutaciones del SDHB producen metástasis. En cambio, el PGL1 raramente presenta un grado de malignidad, éste puede alcanzar menos de un 5%. El PGL1 se asocia primariamente con PGL del cuello y de la cabeza (también conocido como tumor glómico), con el 68% de la penetrancia de este fenotipo, a la edad de los 40 años.
El gen SDHD es impreso en la madre (silenciado) y el tumor se desarrolla únicamente cuando la mutación es heredada del padre. Las excepciones a estas reglas son muy bajas.
El patrón paterno de herencia también se ha descripto en el gen SDHAF2, responsable de un síndrome muy raro de PGL2. El PGL3 causado por la mutación en el gen SDHC y el PGL5 por mutación en el SDHA, estos dos últimos son extremadamente raros.
Al igual que el PGL1, los PGL2, 3 y 5 presentan un bajo riesgo de malignidad. Los PGL torácicos y abdominales se manifiestan en el 69% de los individuos a los 60 años.
Las mutaciones germinales de la TMEM127 no han sido asociadas a un síndrome específico hasta el momento, pero producen en su mayoría Feo bilaterales. Al igual que aquellos que portan el gen MAX, no se ha descripto hasta el momento un síndrome específico. Aunque también frecuentemente se presentan como Feo bilaterales10.
La enfermedad de VHL es la mayor causa de tumor cromafín diagnosticada en la niñez. El Feo o PGL aparece entre el 10-20% de los pacientes con VHL, especialmente en los individuos con VHL2 y con una mutación sin sentido de los genes supresores del tumor de la enfermedad de VHL23,24.
La mayoría de los VHL asociados con Feo o PGL son funcionales, pero también han sido descriptos tumores parasimpáticos de PGL no funcionantes.
Hay un patrón específico de riesgo para el desarrollo del Feo en la NEM 2, la activación en la mutación de los exones 8, 10, 11, 13 y 14 (NEM 2 A) y de los exones 14-16 (NEM 2 B) de los protooncogenes pueden causar un Feo en hasta un 50% de los sujetos afectados. Además, las mutaciones somáticas del RET pueden ser detectadas en el 20% de los Feo esporádicos25-27. La NEM 2 ha sido asociada muy raramente con un PGL parasimpático.
Con todo ello se puede concluir que el test genético28-30, bajo una adecuada supervisión de un grupo de genética, debe realizarse en todos los niños que se presentan con Feo o PGL11,15,31-33. En primer lugar, priorizando el estudio de los genes asociados a VHL. La evaluación de las mutaciones germinales del protooncogen RET sólo es recomendada en los raros casos de niños que se presenten con un Feo aparentemente de aparición esporádica, que tengan un fenotipo bioquímico adrenérgico. Por otra, la neurofibromatosis tipo 1 (NF1) se diagnostica clínicamente.
Feocromocitoma maligno
La gran mayoría de los Feo/PGL son tumores benignos, pero aproximadamente un 12% son malignos en la población pediátrica. Los registros tumorales británicos muestran una incidencia de Feo maligno en la niñez de 0,02 por millón por año31, mientras que otros centros informan un porcentaje tan alto como 46%1,34.
Ningún perfil histológico o inmunohistoquímico es capaz de predecir el potencial metastásico, incluyendo la atipia citológica extrema, la invasión capsular o vascular o áreas remanentes de neuroblastoma1. Las características que más orientan a una malignidad sin la localización extra adrenal son: la necrosis del tumor confluente, la ausencia de glóbulos hialinos, el elevado índice proliferativo, un tamaño mayor a 5 cm, entre otros35.
Se encuentran disponibles scores para determinar malignidad, incluyendo al score de PASS (estratificación del Feo en su localización adrenal) del año 2002 y el subsiguiente score del año 2005 que incorporó las características bioquímicas del tumor36,37.
La malignidad del tumor se establece únicamente ante la presencia de metástasis (MTS) a distancia, en sitios donde normalmente no hay tejido de paraganglia (ejemplo: nódulos linfáticos, hígado, pulmones y hueso). El riesgo de transformación maligna es mayor para el PGL extra adrenal simpático que para el Feo o los PGL no secretantes.
Aunque los PGL del cuello y de la cabeza son en su mayoría no malignos, pueden presentar una alta morbilidad debido al crecimiento local y por la infiltración de las estructuras normales por la masa tumoral.
Presentación clínica
La clínica dependerá de la funcionalidad o del predominio simpático del Feo/PGL que se debe a las diferencias en la secreción de catecolaminas y de la sensibilidad individual a las mismas. Menos frecuentemente, la sintomatología puede deberse al efecto de masa tumoral (por ejemplo dolor), y diagnosticarse por un hallazgo incidental de imágenes o debido a un screening por uno de los síndromes hereditarios discutidos anteriormente31,38.
Los signos y los síntomas del tumor simpático cromafín incluyen HTA sostenida o episodios paroxísticos. La clásica triada de cefalea, palpitaciones y diaforesis de aparición paroxística, palidez, hipotensión ortostática y síncope, temblor, ansiedad.
Otros síntomas que no son específicos pero pueden aparecer son: visión borrosa, dolor abdominal, diarrea u otros síntomas gastrointestinales, pérdida de peso, hiperglucemia, poliuria, polidipsia, fiebre, problemas de conducta o dificultad para realizar las tareas habituales.
La hematuria o los síntomas paroxísticos que ocurren durante la micción pueden observarse en los casos de PGL localizado en vejiga.
Las complicaciones del exceso de catecolaminas puede incluir: crisis hipertensiva, cardiomiopatía, pancreatitis, accidente cerebrovascular (ACV), convulsiones, hasta falla orgánica y muerte39-41.
Los síntomas parasimpáticos de los tumores no secretantes incluyen: pérdida auditiva, tinitus y otros síntomas secundarios por efecto de masa.
Debido al origen neuroendocrino de los tumores, éstos raramente pueden co-secretar otras hormonas, generando un cuadro clínico de exceso hormonal ectópico, tal como es el gigantismo (GHRH), el síndrome de Cushing (ACTH), la hipercalcemia (PTH), el síndrome de secreción inadecuada de hormona antidiurética, la diarrea (péptido intestinal vasoactivo).
El Feo/PGL diagnosticado durante un estudio por un screening familiar es generalmente asintomático42,43. Aunque esta última presentación clínica se está tornando cada vez más frecuente, no hay aún un consenso sobre que debe realizarse en estos pacientes con tumores pequeños asintomáticos, especialmente en aquellos con bajo riesgo de malignidad.
Las manifestaciones cardiológicas pueden ser: dolor precordial, arritmias, hipertrofia cardíaca, shock cardiogénico, ACV, edema agudo de pulmón, infarto agudo de miocardio (IAM) sin enfermedad coronaria. La HTA, como se mencionó previamente, puede presentarse con crisis o sin ella; también, HTA gestacional o paroxística. Otras veces puede observarse hipotensión ortostática.
Crisis de hipertensión arterial
Las crisis hipertensiva es una de las emergencias más frecuentes en el Feo. La crisis puede estar acompañada de cefalea, trastornos visuales, arritmias cardíacas, encefalopatía, IAM, insuficiencia cardíaca o ACV. Es crucial que el tratamiento antihipertensivo apropiado se instaure lo antes posible. Las drogas que se recomiendan para el tratamiento de la crisis hipertensiva son fenoxibenzamina (vía oral: 10 mg/día y puede aumentarse a 100 mg/día) y fentolamina (vía endovenosa o intramuscular). Generalmente, se administra en bolo intravenoso de 2,5 a 5 mg, dando 1 mg por minuto. Si es necesario se puede repetir cada 5 minutos hasta que la HTA sea controlada. También, puede ser administrada en infusión continua (100 mg de fentolamina en 500 ml de dextrosa al 5%). Durante la infusión, debe registrarse el valor de la presión arterial. Sin embargo, en la actualidad la fentolamina tiene una limitada disponibilidad. La administración intravenosa de nitroprusiato, nicardipina o fenoldopam se utilizan más frecuentemente para el tratamiento de la crisis hipertensiva. Si la HTA es menos severa puede utilizarse bloqueantes del receptor alfa tal como la doxazocina, iniciando una dosis de 4 mg hasta alcanzar los 16 mg/día.
El uso de tratamiento con bloqueantes cálcicos orales e intravenosos, perioperatorio, tal como la nicardipina, también puede ser efectivo. Sin embargo, hay que tener en cuenta que no puede usarse cualquier bloqueante cálcico: el diltiazem no previene el aumento de la presión arterial durante la cirugía del Feo, y el verapamilo puede asociarse con la aparición de un edema en el postquirúrgico.
El uso de beta bloqueantes, como: propanolol, esmolol, atenolol y metoprolol, debe usarse sólo después de haber iniciado el tratamiento con bloqueantes alfa (véase tratamiento).
Diagnóstico
El diagnóstico del Feo/PGL ha sido simplificado gracias a los avances en los estudios para detectar y cuantificar los niveles de catecolaminas y sus metabolitos tanto en sangre como en orina. En la actualidad el test diagnóstico de elección es la medición de las metanefrinas plasmáticas y/o urinarias (metanefrina y normetanefrina), las cuales son altamente sensibles alcanzando una sensibilidad del 100% para el diagnóstico de tumores cromafines simpáticos (Tabla 1). Los métodos que utilizan la espectrometría de masa para medir las metanefrinas plasmáticas libres parecen ser superiores44,45.
Tabla 1. Diagnóstico bioquímico de Feo/PGL Sensibilidad y especificidad de las pruebas de laboratorio para Feo/PGL
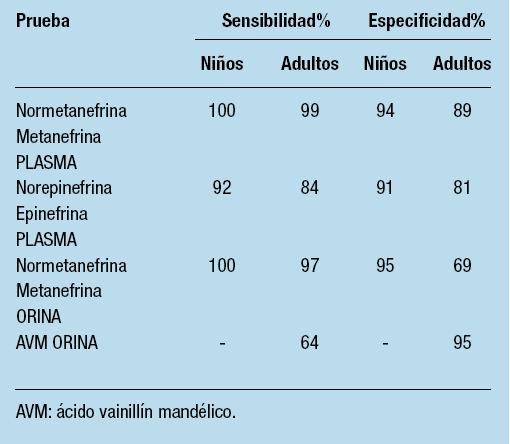
La alta sensibilidad del test de metanefrinas está basada en el hecho de que hay un metabolismo intratumoral de catecolaminas, de noradrenalina (norepinefrina) a normetanefrinas y de adrenalina (epinefrina) a metanefrina, un proceso que sucede independientemente de la liberación de catecolaminas que puede aparecer en forma intermitente o en bajo índice46. Un aumento de 4 veces los valores de referencia se asocian a un 100% de probabilidad de estar ante la presencia de un tumor secretante de catecolaminas. Ciertas drogas interfieren con la prueba por lo cual deberían ser suspendidas antes de realizar el estudio, tal como: paracetamol, los descongestivos nasales, los antidepresivos tricíclicos, fenoxibenzamina entre otros (Tabla 2).
Tabla 2. Causas de falsos positivos

Además, para evitar falsos positivos generados por el ejercicio o los procedimientos que generan estrés, las metanefrinas se deberían dosar en la posición supina luego de 30 minutos de reposo. Las restricciones alimentarias no son necesarias, excepto que se sospeche de un tumor secretante de dopamina o que sólo se realice el dosaje de normetanefrinas desconjugados o se dose ácido vainillín mandélico (AVM)47.
Los tumores funcionales pueden ser subclasificados en noradrenérgicos o adrenérgicos, basados en los patrones de liberación de catecolaminas. Los Feo o PGL noradrenérgicos se caracterizan por producir noradrenalina y normetanefrina48, como se observa en la enfermedad de VHL y en muchos de los síndromes asociados a PGL familiar.
Los tumores adrenérgicos secretan tanto adrenalina como noradrenalina y sus respectivos metabolitos; esto frecuentemente es hallado en el Feo que aparece esporádicamente o dentro del contexto clínico de la NEM 2 o de la NF149.
Una de las razones en cuanto a la secreción de catecolaminas es la disminuida expresión de la N-metiltransferasa feniletanolamida, enzima que convierte a la noradrenalina en adrenalina en los tumores de la enfermedad de VHL, en comparación a aquellos que la aumentan como la NEM 2.
Muy raramente los tumores secretan preferentemente dopamina. El tumor secretante de dopamina debe ser considerado en pacientes normotensos, que presentan una masa que hace sospechar un Feo o PGL, en cuyo caso la dopamina y sus metabolitos deben ser medidos.
La cromogranina A es la principal proteína secretada a partir de los gránulos cromafines de la matriz soluble, siendo un marcador tumoral efectivo que se correlaciona con el tamaño tumoral y el potencial maligno, y aumenta la sensibilidad del test diagnóstico y del seguimiento a largo plazo.
En aquellos pacientes con síntomas paroxísticos y niveles normales de metanefrinas, la confirmación diagnóstica puede ser mejor alcanzada a través del dosaje de las metanefrinas fraccionadas y de las catecolaminas durante y justo luego del evento clínico.
En cuanto a los test de estimulación con glucagon y de la supresión con clonidina, han sido ampliamente estudiados en el diagnóstico de tumores secretantes de catecolaminas, pero son infrecuentemente utilizados, y en el caso de la estimulación con glucagon, han sido abandonados debido a la insuficiente sensibilidad diagnóstica.
Estudios de imágenes
Una vez que se estableció que hay un exceso de catecolaminas, para la localización del tumor se utilizan imágenes.
Los test iniciales para realizar el diagnóstico son la tomografía computada (TAC) (Figura 4) y la resonancia magnética nuclear (RMN) (Figura 5).
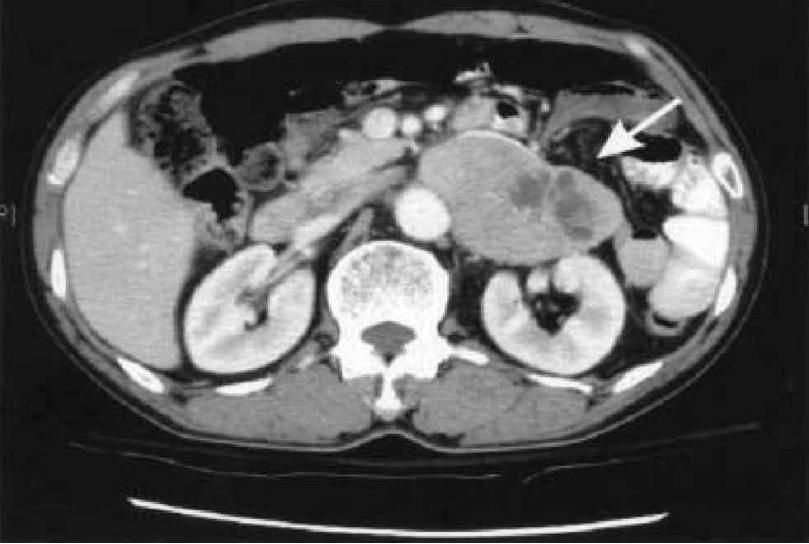
Figura 4. Tomografía axial computada que muestra una masa adrenal izquierda con áreas de necrosis y áreas de hemorragia.
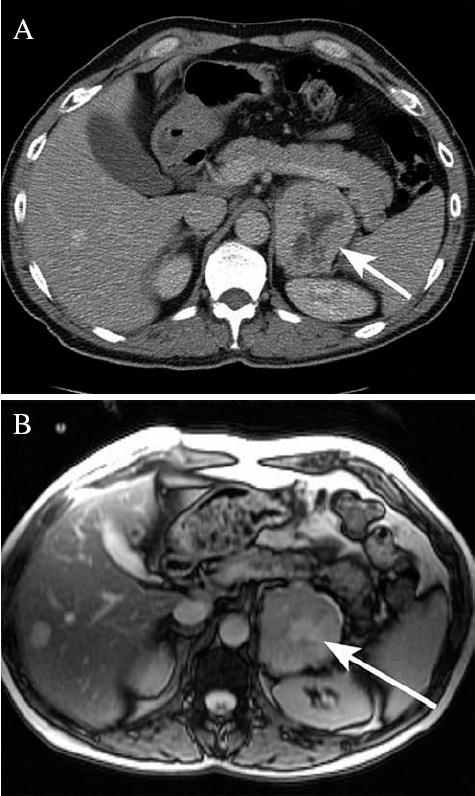
Figura 5. Tomografía axial computada (A) y resonancia magnética nuclear (B) de un mismo paciente portador de un feocromocitoma, masa adrenal izquierda con áreas de necrosis central (área de hiperintensidad).
Los Feo/PGL son tumores vasculares que contienen áreas de necrosis, quísticas o áreas de hemorragias que en la RMN se observan como áreas de hiperintensidad en la secuencia de T2 (Figura 6).
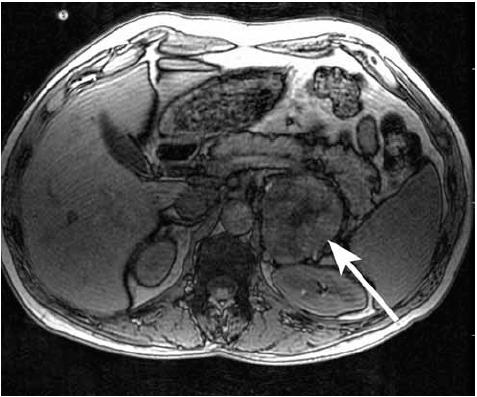
Figura 6. Resonancia magnética nuclear de un paciente portador de un feocromocitoma. Masa adrenal izquierda heterogénea con áreas de necrosis central (área de hiperintensidad).
Los test funcionales utilizan la centellografía nuclear con la metaiodobenzilguanidina (MIBG). Esta prueba es altamente específica para confirmar la secreción natural del tumor de catecolaminas (Figura 7). Antes de realizar esta prueba con MIBG hay que asegurar que el paciente no haya ingerido descongestivos, bloqueantes cálcicos o labetalol, drogas que disminuyen la captación de MIBG50.

Figura 7. Centellografía nuclear con la metaiodobenzilguanidina (MIBG). A: captación del radioisótopo cuatro horas después de la administración de MIBG. B: imágenes fusionadas con SPECT-CT que muestran la concentración focal de MIBG (flecha) dentro de la glándula suprarrenal izquierda (feocromocitoma).
Debido a que el test con MIBG no es 100% sensible, otras modalidades con imágenes nucleares deben ser consideradas, como la centellografía destinada al receptor de la somatostatina, tomografía de emisión de positrones (PET) para fluorodihidrofenilalanina (DOPA) (Figura 8), para fluorodopamina (FDA), fluorodeoxiglucosa (FDG), epinefrina, hidroxiepinefrina. Hay que tener en cuenta que no todos los centros tienen disponibilidad para realizar estos estudios (Tabla 3).
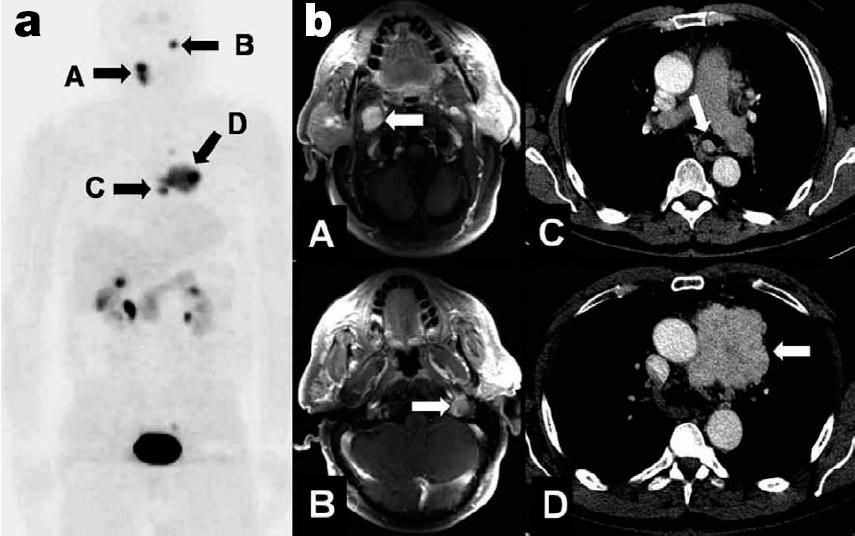
Figura 8. Síndrome paraganglionar tipo 1. a. Imagen de tomografía de emisión de positrones con 18-F-DOPA con al menos 4 tumores identificados. b. Imágenes de resonancia magnética nuclear de los 4 tumores hallados en a.
Tabla 3. Estudios de imágenes para el feocromocitoma Diagnóstico de localización

Estadificación y pronóstico
El pronóstico para un tumor resecado en su totalidad, especialmente para un Feo, es excelente.
La expectativa de vida para una enfermedad maligna dependerá del sitio de localización de las metástasis, con una sobrevida menor a 5 años en los pacientes que presentan metástasis a nivel pulmonar o hepático, y mayor sobrevida para los que presentan metástasis ósea. La sobrevida a los 5 años varía de un 34% a un 60%, aunque puede ser menor, llegando casi a un 25%, en algunas series de 15 años o más51.
Hasta la actualidad no se dispone de un sistema de estadificación para el Feo/PGL maligno.
La influencia genética y la bioquímica nos permiten dividir al Feo/PGL en dos grupos:
- Grupo 1 incluye al síndrome de VHL y a los tumores que tienen una mutación en la SDH, y se caracterizan por un aumento de la noradrenalina y en algunos casos de la dopamina.
- Grupo 2 incluye al RET, NF1, TMEM127 y el MAX, caracterizados por un aumento de adrenalina y a veces de noradrenalina.
Tratamiento
Tratamiento médico
La medicación preparatoria para la cirugía, una vez que se halla confirmado bioquímicamente el diagnóstico de un Feo/PGL simpático, debe iniciarse 1 a 2 semanas antes de la cirugía. Esta medicación preparatoria se utiliza para minimizar las complicaciones que pueden ocurrir ante la liberación aguda de catecolaminas, durante la inducción anestésica y la manipulación del tumor52. No hay un algoritmo universal para este tratamiento médico; sin embargo, el bloqueo adrenérgico es generalmente el tratamiento de elección, utilizándose la fenoxibenzamina fenoxibenzamina (10 mg/día vía oral). Los efectos adversos de la fenoxibenzamina incluyen taquicardia, congestión nasal e hipotensión ortostática. Los bloqueantes selectivos tales como prazocina o doxazocina y los bloqueantes de los canales de calcio, tal como nifedipina o nicardipina también pueden ser utilizados. El tratamiento con los bloqueantes alfa mejoran los síntomas, disminuyen la presión arterial y aumenta el volumen intravascular.
Debido a que la fenoxibenzamina es una medicación de larga vida media, puede aumentar el riesgo posoperatorio de hipotensión. La metirosina es un inhibidor competitivo de la tirosina hidroxilasa, limitando la síntesis de catecolaminas, también puede ser utilizado como tratamiento prequirúrgico. Sin embrago, debido a la gran cantidad de efectos adversos que puede presentar (especialmente diarrea, sedación y manifestaciones extrapiramidales), no se recomienda de manera rutinaria.
Debido a la hipotensión ortostática que pueden generar estos fármacos, se recomienda iniciar el tratamiento a dosis bajas e ir incrementándola hasta alcanzar un adecuado nivel de presión arterial.
Una vez que se ha completado el bloqueo alfa, se inicia con agentes beta bloqueantes (tales como el propanolol, atenolol o metoprolol) para controlar la taquicardia refleja53.
Los beta bloqueantes no deben ser utilizados nunca como monodroga, debido a la posibilidad de empeorar los síntomas y la HTA, a causa del efecto que las catecolaminas puedan ejercer sobre los receptores adrenérgicos.
Unos días previos a la cirugía, se recomienda una sobrecarga de sal, para evitar la severa hipotensión arterial que pueda ocurrir en el postoperatorio. En algunos centros, internan al paciente previamente para medicarlo en forma endovenosa, previamente, a la resección del Feo/PGL.
Tratamiento quirúrgico
La resección quirúrgica es el tratamiento pilar para el Feo/PGL (Tabla 4).
Tabla 4. Tratamiento de Feo/PGL

La adrenalectomía puede realizarse utilizando la vía laparoscópica, tanto transperitoneal como retroperitoneal54. La biopsia preoperatoria no está indicada por el riesgo potencial que puede englobar. La laparotomía se debe plantear cuando se sospecha que el Feo es de gran tamaño o es maligno, según la sospecha clínica y/o de imágenes. Además del tratamiento quirúrgico convencional, la tecnología robótica está siendo cada vez más utilizada. Esta última técnica ha demostrado un elevado éxito y una reducida mortalidad en comparación a la laparoscopía.
En cambio, el tratamiento óptimo para un paciente con un Feo bilateral producido por un síndrome hereditario es controversial, debido a su resultado postquirúrgico. Aunque la adrenalectomía bilateral teóricamente elimina el riesgo de recurrencia tumoral, genera una dependencia de por vida para los esteroides y el riesgo de la potencial fatalidad por una crisis addisoniana, aunque es poco frecuente en la actualidad.
Si se desea realizar un procedimiento bilateral, se recomienda realizar la retroperitoneoscopía posterior, ésta ofrecería un beneficio adicional y reduciría el tiempo quirúrgico. Se debe plantear el procedimiento de conservación cortical, especialmente ante población joven, para evitar la necesidad de reemplazo hormonal de por vida, que se requeriría si se realizara una adrenalectomía bilateral como se mencionó anteriormente.
Para poder constatar que haya sido un procedimiento de conservación cortical exitoso, debe realizarse una TAC con imágenes de reconstrucción vascular, y evidenciarse la corteza. Es extremadamente difícil preservar la porción vascular de la corteza adrenal, la cual debe ser suficiente para evitar la dependencia corticoesteroidea, sin dejar cierto resto de la médula, la cual es el riesgo para una recurrencia del Feo, ante la persistencia de un remanente. Se dispone de datos limitados sobre la recurrencia de un Feo, luego de una adrenalectomía cortical, siendo aproximadamente del 10% al 38%55. La recurrencia de un Feo no necesariamente es en el mismo lugar del tumor previo.
En pacientes con NEM 2 y con síndrome de VHL, la adrenalectomía cortical debe plantearse como elección, pues el 65% de los pacientes que se somete a este procedimiento no dependerán de por vida de los corticoides. Esto se ha demostrado a largo plazo y el riesgo de una recurrencia en estos casos es de un 10%.
La decisión de cuanta glándula adrenal podrá preservarse in situ, dependerá de cómo el cirujano encuentra en el acto quirúrgico a la glándula y su vascularización.
Aunque la laparoscopía ha sido considerada como segura en pacientes con Feo benigno, hay una limitada experiencia con los tumores metastásicos. El uso de laparoscopia en pacientes con tumor primario de la glándula adrenal es controversial, debido a que la laparoscopía es inferior para la obtención de muestras y el riesgo de ruptura tumoral, es mayor en aquellos de gran tamaño.
En cuanto al PGL, la conducta quirúrgica dependerá del sitio donde se encuentre el tumor, en ciertos casos podrá aplicarse también la laparoscopía.
Es importante que el anestesiólogo esté familiarizado con el manejo perioperatorio e intraoperatorio del Feo/PGL, debido a que la presión arterial puede variar ampliamente al igual que la aparición de arritmias, especialmente, en el período de la inducción anestésica y del manipuleo del tumor. En el posoperatorio, es fundamental estar alerta para evitar dos principales complicaciones: la hipoglucemia y la hipotensión arterial.
Tratamiento de la metástasis
Pocas terapias sistemáticas son planteadas para el tratamiento de un Feo más agresivo y de los PGL. Se utiliza en los PGL malignos un tratamiento con agentes metabólicos, tales como aquellos que transportan metaiodobencilguanidina o análogos de la somatostatina. Cuando progresa la enfermedad se utilizan agentes citostáticos tales como ciclofosfamida, dacarbazina, vincristina y doxorubicina, siendo en su mayoría un tratamiento paliativo más que curativo. Aproximadamente, el 31% de los pacientes tratados con quimioterapia demuestran una mejoría en cuanto a su hipertensión arterial, dolor y tamaño tumoral, y por ende, una franca mejoría de la sobrevida.
Se desconoce cuáles son los factores predictores de una adecuada respuesta a la quimioterapia.
Hay drogas en estudio tales como ultratrace (I131) iobenguane que ha demostrado una mejoría en la presión arterial y una reducción en el tamaño tumoral. También terapias moleculares están siendo prometedoras para este tipo de patologías.
Debido a que un Feo/PGL es altamente vascular, sunitinib (un inhibidor de las kinasas con propiedades antiangiogénicas) puede ser un agente terapéutico prometedor.
El feocromocitoma en su presentación como miocardiopatía
Como vimos anteriormente, el Feo puede presentarse típicamente con cefaleas paroxísticas, nauseas, palpitaciones e hipertensión arterial. Pero en algunas ocasiones, la secreción catecolaminérgica del Feo induce un síndrome clínico que imita a la cardiomiopatía por estrés (Tako-tsubo), es decir, es capaz de producir una cardiomiopatía reversible. Gracias al mayor uso de las imágenes cardíacas, se ha demostrado que el Feo produce patrones específicos en la motilidad cardíaca, cuando afecta el miocardio ante el exceso catecolaminérgico.
Desde el año 1969, gracias a los aportes de Wiswell, se ha visto que el Feo es capaz de producir una miocardiopatía reversible. Estudió un grupo de tan sólo 13 pacientes con Feo que habían manifestado sintomatología cardíaca56, un total de 6 pacientes habían presentado cambios electrocardiográficos, incluyendo supradesnivel del segmento ST, infradesnivel del segmento ST e inversión de la onda T, y todos resueltos luego de la extracción tumoral (Figura 9). Ante estos hallazgos concluyó que el Feo podía presentar una forma reversible de miocardiopatía, sin obstrucción coronaria causado únicamente por el incremento exagerado de catecolaminas.
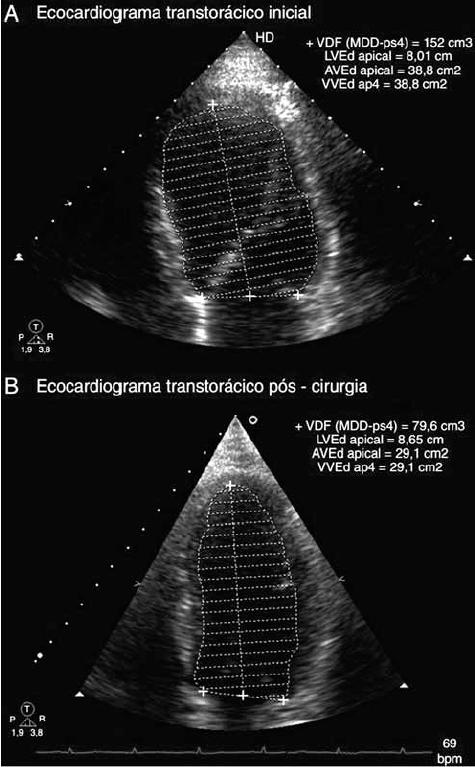
Figura 9. Ecocardiograma bidimensional transtorácico en vista apical de 4 cámaras en telediástole. A: antes de la cirugía. B: pos cirugía, donde evidencia la reducción del volumen telediastólico del ventrículo izquierdo.
Ante la aparición de las imágenes cardíacas a fines de los años 1980, observó que el Feo se acompañaba de una hipoquinesia o aquinesia apical con hiperquinesia basal.
En el caso de la miocardiopatía de Tako-tsubo, puede ser clasificada como típica o atípica o invertida. Esta última se caracteriza por presentar hiperquinesia de los segmentos apicales con hipoquinesia de los segmentos basales o mediales, esta forma puede ser más frecuente de lo que se pensaba, con una relación aproximada del 60/40, respectivamente.
En cuanto a la miocardiopatía reversible del Feo, ésta puede ser descripta como variante típica o invertida del Tako-tsubo. Este cuadro se presenta como disfunción diastólica del ventrículo izquierdo, sin evidencia de lesiones coronarias.
La clásica presentación de la cardiomiopatía por estrés, usualmente, es gatillada por un severo estrés emocional o físico, produciendo una severa hipoquinesia apical y una hiperquinesia basal que imita un síndrome coronario agudo57.
Las características clínicas del síndrome de Tako-tsubo, según The Mayo Clinic, incluyen:
1) Disquinesia, aquinesia o hipoquinesia transitoria del ventrículo izquierdo no representada por una sola lesión epicárdica.
2) Ausencia de lesión coronaria obstructiva.
3) Cambios electrocardiográficos tales como elevación del segmento ST y/o inversión de la onda T o elevación de la troponina.
4) Ausencia de trauma endocraneano, hemorragia intracraneana, enfermedad coronaria, Feo, miocarditis y miocardiopatía hipertrófica.
Si el Feo o el PGL no se diagnostican, la miocardiopatía por estrés puede aparecer en forma recurrente en el mismo individuo. En este contexto también pueden observarse taquicardias ventriculares.
Por décadas, se ha conocido que el Feo es causante de una miocardiopatía reversible; el aumento en el reconocimiento de la miocardiopatía de Tako-tsubo hace que, más frecuentemente, se encuentre una conexión entre ambas patologías, sosteniendo un rol de las catecolaminas a través de una hibernación miocárdica o una toxicidad miocárdica directa. Cada vez con mayor frecuencia, se publican casos de Feo que se presentan como un patrón invertido de Tako-tsubo, pudiendo reflejar una nueva asociación fisiopatológica.
También, el Feo puede presentarse como una hipertrofia del ventrículo izquierdo a nivel apical similar a la miocardiopatía hipertrófica apical. En el trazado electrocardiográfico pueden observarse ondas T negativas picudas (1,0 mV), y en el ecocardiograma y en la angiografía se observa una hipertrofia ventricular localizada a nivel apical, que al año de la cirugía de adrenalectomía evoluciona con regresión de la misma y normalización electrocardiográfica. Este hallazgo enfatiza el rol de las catecolaminas en la hipertrofia apical y la potencial reversibilidad del daño de órgano blanco con el Feo58. Los posibles mecanismos fisiopatológicos de la disfunción miocárdica son: disfunción microvascular de las arterias coronarias, espasmo de los vasos epicárdicos, afectación en el metabolismo lipídico, disfunción miocárdica mediada por catecolaminas.
La porción apical del miocardio parecería ser más vulnerable al incremento de las catecolaminas circulantes debido a la variada distribución de la inervación simpática y a la disímil densidad de los nervios simpáticos en el corazón. La variación de los segmentos comprometidos, en los pacientes con excesivas concentraciones de catecolaminas, puede sugerir una diferente susceptibilidad a la estimulación del sistema simpático de un individuo a otro59.
Hipotensión arterial y shock
Raramente, se observa hipotensión arterial severa o shock en pacientes con Feo. Este cuadro puede estar precedido por paroxismos de hipertensión arterial.
En menos del 2% de los pacientes con Feo se observa el shock, y en algunos pacientes se puede manifestar el cuadro como "pseudoshock". En este caso, la resistencia vascular periférica no es medible debido a una vasoconstricción periférica extrema, mientras que la presión arterial central es muy elevada.
En algunos pacientes que presentan shock, el mismo se puede acompañar de dolor abdominal severo, edema pulmonar, diaforesis y cianosis.
En mucho de los pacientes que presentan hipotensión o shock, el tumor parecería secretar epinefrina.
El mecanismo por el cual pueden producirse ambos cuadros no está completamente resuelto. La hipovolemia intravascular sumado a la disminución del volumen minuto podrían ser los factores probables como contribuyentes al shock del Feo. La hipocalcemia puede ser un factor patogénico importante, que genera disminución en la contractilidad cardíaca y que contribuye a la aparición del shock cardiogénico en los pacientes con Feo secretante de epinefrina.
En los casos de necrosis hemorrágica, en aquellos tumores de gran tamaño, la abrupta interrupción en la secreción de catecolaminas puede producir una repentina hipotensión arterial o shock.
Conclusión
El 85% de los casos de un Feo se encuentran en la médula adrenal o suprarrenal y un 15% en el tejido cromafín extra-adrenal60. Presentando una baja prevalencia (entre el 0,05-0,1%), siendo el 50% de los pacientes sintomático. El principal problema clínico con el Feo/PGL es la dificultad para realizar el diagnóstico, debido a su extrema variabilidad en su presentación clínica. No hay signos o síntomas que sean específicamente suficientes como para permitir el diagnóstico. Se sabe, que muchos de los casos de Feo/PGL son detectados en imágenes como incidentalomas a nivel adrenal, abdominal, torácico o cervical.
La excesiva secreción de catecolaminas es la causante de la florida sintomatología. Las manifestaciones son diversas y el tumor puede imitar una gran variedad de enfermedades, produciendo frecuentemente un diagnóstico tardío o erróneo, por ello lleva el nombre del "gran imitador"61.
La estimulación de los receptores alfa genera hipertensión arterial, aumento de la contractilidad cardíaca, glucogenolisis, gluconeogénesis, relajación intestinal. La estimulación de los receptores beta genera aumento de la frecuencia cardíaca y de la contractilidad. La secreción excesiva de catecolaminas puede finalizar con un cuadro de miocardiopatía reversible, acompañada o no de insuficiencia cardíaca.
Se sospecha que un paciente es portador de un Feo ante la presencia de palpitaciones, cefalea intensa, nauseas, sudoración, temblor, palidez, HTA refractaria, diabetes tipo II atípica, síndromes familiares que predisponen a Feo/PGL, historia familiar de Feo, incidentaloma en suprarrenales, HTA en jóvenes, miocardiopatía dilata idiopática.
Si es correctamente diagnosticado y tratado, el Feo puede ser curado; en cambio, si no se diagnostica o no se trata adecuadamente presenta una elevada morbi-mortalidad.
El diagnóstico precoz es esencial para el tratamiento efectivo que, generalmente es la resección quirúrgica.
Se recomienda realizar dosaje en el laboratorio de metanefrinas plasmáticas y urinarias. En los últimos 10 años, los estudios genéticos han demostrado que el Feo/PGL son los mayores tumores hereditarios hasta el momento, entre el 30%-40% de los tumores son genéticamente heredados.
La genética tiene un impacto relevante en cuanto al manejo clínico de los pacientes afectados. De hecho, la genética influye en el patrón secretante, el perfil histoquímico, la presentación clínica, las características metabólicas, el riesgo de malignización, los mecanismos intracelulares comprometidos en el desarrollo tumoral.
En el futuro, la genética podrá ser una herramienta para el tratamiento en los pacientes que presentan metástasis de esta enfermedad.
1. Tischler AS. Pheochromocytoma and extra-adrenal paraganglioma: updates. Arch Pathol Lab Med 2008;132(8):1272-1284. [ Links ]
2. Goodman MT, Gurney JG, Smith MA, Olshan AF. Sympathetic nervous system tumors. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. NIH Pub. No. 99-4649. Bethesda, MD: National Cancer Institute1999; 65-72. [ Links ]
3. Wyszyñska T, Cichocka E, Wieteska-Klimczak A, Jobs K, Januszewicz P. A single pediatric center experience with 1025 children with hypertension. Acta Paediatr 1992;81:244-246. [ Links ]
4. Young Jr WF. Endocrine hypertension. In: Kronenberg HM, Melmed S, Polonsky KS, Larsen PR, eds. Williams textbook of endocrinology. 11th ed. Philadelphia: Saunders Elsevier 2008;505-537. [ Links ]
5. Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C, Landes C, McPartland JL, Moores C, Losty PD, Didi M. Phaeochromocytoma in children. Arch Dis Child 2008;93:899-904. [ Links ]
6. Timmers HJ, Pacak K, Huynh TT, Abu-Asab M, Tsokos M, Merino MJ, Baysal BE, Adams KT, Eisenhofer G. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J Clin Endocrinol Metab 2008;93:4826-4832. [ Links ]
7. Linnoila RI, Keiser HR, Steinberg SM, Lack EE. Histopathology of benign versus malignant sympathoadrenal paragangliomas: clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol 1990;21:1168-1180. [ Links ]
8. Juárez D, Brown RW, Ostrowski M, Reardon MJ, Lechago J, Truong LD. Pheochromocytoma associated with neuroendocrine carcinoma. A new type of composite pheochromocytoma. Arch Pathol Lab Med 1999;123:1274-1279. [ Links ]
9. Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C. Germline mutations in nonsyndromic pheochromocytoma. N Engl J Med 346(19):1459-1466. [ Links ]
10. Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul JL, Strompf L, Schlumberger M, Bertagna X, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005;23:8812-8818. [ Links ]
11. Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, Pignataro V, Bernini G, Giache V, Bacca A, Biondi B, Corona G, DiTrapani G, Grossrubatscher E, Reimondo G, Arnaldi G, Giacchetti G, Veglio F, Loli P, Colao A, Ambrosio MR, Terzolo M, Letizia C, Ercolino T, Opocher G. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab 2009;94:1541-1547. [ Links ]
12. Kimura N, Miura Y, Nagatsu I, Nagura H. Catecholamine synthesizing enzymes in 70 cases of functioning and non-functioning phaeochromocytoma and extra-adrenal paraganglioma. Virchows Arch A Pathol Anat Histopathol 1992;421:25-32. [ Links ]
13. Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K. Pheochromocytomas in von Hippelhyphen Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001;86:1999-2008. [ Links ]
14. Jiménez C, Cote G, Arnold A, Gagel RF. Should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? J Clin Endocrinol Metab 2006 91:2851-2858. [ Links ]
15. Dworakowska D, Grossman AB. Are neuroendocrine tumors a feature of tuberous sclerosis? A systematic review. Endocr Relat Cancer 2009;16:45-58. [ Links ]
16. Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells Jr SA, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658-5671. [ Links ]
17. Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib H, Moley JF, Pacini F, Ringel MD, Schlumberger M, Wells Jr SA. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009;19:565-612. [ Links ]
18. Erlic Z, Neumann HP. Familial pheochromocytoma. Hormones 2009;8:29-38. [ Links ]
19. Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer 2007;14:935-956. [ Links ]
20. Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, Feunteun J, Pouysségur J, Richard S, Gardie B. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med 2008;359:2685-2692. [ Links ]
21. Carney JA. Carney triad: a syndrome featuring paraganglionic, adrenocortical, and possibly other endocrine tumors. J Clin Endocrinol Metab 2009; 94:3656-3662. [ Links ]
22. Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 1999;74:543-552. [ Links ]
23. Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, Moore AT. Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet 1996;33:328-332. [ Links ]
24. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. Von Hippel-Lindau disease. Lancet 2003;361:2059-2067. [ Links ]
25. Beldjord C, Desclaux-Arramond F, Raffin-Sanson M, Corvol JC, De Keyzer Y, Luton JP, Plouin PF, Bertagna X. The RET protooncogene in sporadic pheochromocytomas: frequent MEN 2-like mutations and new molecular defects. J Clin Endocrinol Metab 1995;80:2063-2068. [ Links ]
26. Lindor NM, Honchel R, Khosla S, Thibodeau SN. Mutations in the RET protooncogene in sporadic pheochromocytomas. J Clin Endocrinol Metab 1995;80:627-629. [ Links ]
27. Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, Niccoli P, Gaillard D, Chabrier G, Chabolle F, Coupier I, Thieblot P, Lecomte P, Bertherat J, Wion-Barbot N, Murat A, Venisse A, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab 2009;94:2817-2827. [ Links ]
28. Van Baars F, Cremers C, van den Broek P, Geerts S, Veldman J. Genetic aspects of nonchromaffin paraganglioma. Hum Genet 1982;60:305-309. [ Links ]
29. Bleker RJ, Wereldsma JC. Carotid body tumor: familial occurrence. Neth J Surg 1986;38:76-80. [ Links ]
30. Mariman EC, van Beersum SE, Cremers CW, Struycken PM, Ropers HH. Fine mapping of a putatively imprinted gene for familial non-chromaffin paragangliomas to chromosome 11q13.1: evidence for genetic heterogeneity. Hum Genet 1995;95:56-62. [ Links ]
31. Spoudeas HA. Paediatric endocrine tumours. West Sussex, UK: Novo Nordisk: 2005. [ Links ]
32. Armstrong R, Sridhar M, Greenhalgh KL, Howell L, Jones C, Landes C, McPartland JL, Moores C, Losty PD, Didi M. Phaeochromocytoma in children. Arch Dis Child 2008;93:899-90. [ Links ]
33. Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 2003;95:1196-1204. [ Links ]
34. Pham TH, Moir C, Thompson GB, Zarroug AE, Hamner CE, Farley D, van Heerden J, Lteif AN, Young Jr WF. Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics 2006;118:1109-1117. [ Links ]
35. Chrisoulidou A, Kaltsas G, Ilias I, Grossman AB. The diagnosis and management of malignant phaeochromocytoma and paraganglioma. Endocr Relat Cancer 2007;14:569-585. [ Links ]
36. Thompson LD. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002;26:551-566. [ Links ]
37. Kimura N, Watanabe T, Noshiro T, Shizawa S, Miura Y. Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: a clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol 2005;16:23-32. [ Links ]
38. Januszewicz P, Wieteska-Klimczak A, Wyszyñska T. Pheochromocytoma in children: difficulties in diagnosis and localization. Clin Exp Hypertens A 1990;12:571-579. [ Links ]
39. Ein SH, Pullerits J, Creighton R, Balfe JW. Pediatric pheochromocytoma. A 36-year review. Pediatr Surg Int 1997;12:595-598. [ Links ]
40. Sullivan J, Groshong T, Tobias JD. Presenting signs and symptoms of pheochromocytoma in pediatric-aged patients. Clin Pediatr 2005;44:715-719. [ Links ]
41. Bissada NK, Safwat AS, Seyam RM, Al Sobhi S, Hanash KA, Jackson RJ, Sakati N, Bissada MA. Pheochromocytoma in children and adolescents: a clinical spectrum. J Pediatr Surg 2008;43:540-543. [ Links ]
42. Pomares FJ, Canas R, Rodríguez JM, Hernández AM, Parrilla P, Tebar FJ. Differences between sporadic and multiple endocrine neoplasia type 2A phaeochromocytoma. Clin Endocrinol 1998;48:195-200. [ Links ]
43. Walther MM, Reiter R, Keiser HR, Choyke PL, Venzon D, Hurley K, Gnarra JR, Reynolds JC, Glenn GM, Zbar B, Linehan WM. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol 1999;162:659-664. [ Links ]
44. Jong WH, Graham KS, van der Molen JC, Links TP, Morris MR, Ross HA, de Vries EG, Kema IP. Plasma free metanephrine measurement using automated online solid-phase extraction HPLC tandem mass spectrometry. Clin Chem 2007;53:1684-1693. [ Links ]
45. Young Jr WF. Pheochromocytoma in children. UpToDate Online 2008. http://www.uptodate.com/contents/pheochromocytoma-in-children
46. Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab 2002;87:1955-1960.
47. Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, Kimura N, Mannelli M, McNicol AM, Tischler AS. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 2007;3(2):92-102. [ Links ]
48. Lumachi F, Tregnaghi A, Zucchetta P, Cristina Marzola M, Cecchin D, Grassetto G, Bui F. Sensitivity and positive predictive value of CT, MRI and 123I-MIBG scintigraphy in localizing pheochromocytomas: a prospective study. Nucl Med Commun 2006;27:583-587. [ Links ]
49. Berglund AS, Hulthén UL, Manhem P, Thorsson O, Wollmer P-2001 Metaiodobenzylguanidine (MIBG) scintigraphy and computed tomography (CT) in clinical practice. Primary and secondary evaluation for localization of phaeochromocytomas. J Intern Med 2001;249(3):247-251. [ Links ]
50. Shulkin BL, Shapiro B. Current concepts on the diagnostic use of MIBG in children. J Nucl Med 1998;39:679-688. [ Links ]
51. Nomura K, Kimura H, Shimizu S, Kodama H, Okamoto T, Obara T, Takano K. Survival of patients with metastatic malignant pheochromocytoma and efficacy of combined cyclophosphamide, vincristine, and dacarbazine chemotherapy. J Clin Endocrinol Metab 2009;94:2850-2856. [ Links ]
52. Goldstein RE, ONeill Jr JA, Holcomb 3rd GW, Morgan 3rd WM, Neblett 3rd WW, Oates JA, Brown N, Nadeau J, Smith B, Page DL, Abumrad NN, Scott Jr HW. Clinical experience over 48 years with pheochromocytoma. Ann Surg 1999;229:755-764; discussion 764-766. [ Links ]
53. Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab 2007;92:4069-4079. [ Links ]
54. Callender GG, Kennamer DL, Grubbs EG, Lee JE, Evans DB, Perrier ND. Posterior retroperitoneoscopic adrenalectomy. Adv Surg 2009;43:147-157. [ Links ]
55. Asari R, Scheuba C, Kaczirek K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with multiple endocrine neoplasia type 2A. Arch Surg 2006;141:1199-1205. [ Links ]
56. Wiswell JG, Crago RM. Reversible Cardiomyopathy with pheocromocytoma. Trans Am Clin Climatol Assoc 1969;80:185-195. [ Links ]
57. Bernard M, Gold M, Cuoco F. Pheochromocytoma presenting as Recurrent Stress Cardiomyopathy with Multiple Monomorphic Ventricular Tachycardias. The Journal Innovations Cardiac Rhythm Management 2012;3:803-808. [ Links ]
58. Schuiki Er, Jenni R, Amann Fw, Ziegler Wh. A reversible form of apical left ventricular hypertrophy associated with Pheochromocytoma. J Am Soc Echocardiog 1993;6(3):327-31. [ Links ]
59. Pierpont GL, DeMaster EG, Cohn JN. Regional Differences in adrenergic function within the left ventricle. Am J Physiol 1984;246:H824-9. [ Links ]
60. Elder E, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma Syndrome: no longer the 10%tumor.J Surg Oncol 2005;89:193-201. [ Links ]
61. Decourcy JL. Pheocromocytoma. Am J Surg 1953;86:37-44. [ Links ]

